Osseous Metastases of Renal Epithelioid Angiomyolipoma with Dramatic Response Through mTOR Inhibitor Therapy
Received Date: September 28, 2021 Accepted Date: October 28 , 2021 Published Date: November 01, 2021
doi: 10.17303/JCRTO.2021.9.205
Citation:SL Barton (2021) Osseous Metastases of Renal Epithelioid Angiomyolipoma with Dramatic Response Through mTOR Inhibitor Therapy. J Cancer Res Therap Oncol 9: 1-8.
Abstract
Introduction:Epithelioid angiomyolipoma of the kidney is a rare, but potentially malignant variant of angiomyolipoma and difficult to treat in metastatic form.
Case:Five years after nephrectomy due to a kidney tumor, which histologically turned out to be an epithelioid angiomyolipoma, a 70-year-old patient presented with histologically confirmed metastasis in the right ilium and lumbal vertebral body metastases. We started therapy with temsirolimus. The first staging MRI examination after three months showed a very good response of the bone metastases to therapy. Therefore, the therapy was continued, and after 13 months, as of 05/2021, the FDG-PET/CT showed complete remission with only avital lesions left.
Conclusion:This case report describes the dramatic response of osseous metastases from an epithelioid angiomyolipoma of the kidney to mTOR inhibitor therapy with temsirolimus.
Keywords:Epithelioid Angiomyolipoma; mTOR Inhibitor; Temsirolimus
List of Abbreviations:ADC: Apparent Diffusion Coefficient; AML: Angiomyolipoma; CT: Computed Tomography; Dr. med: Doctor medicine; Dr. rer. Nat: Doctor rerum naturalium; DWI: Diffusion-Weighted Imaging; EAML: Epithelioid angiomyolipoma: FDG-PET/CT: fluorodeoxyglucose (FDG)-positron emission tomography/ Computed tomography; HMB45: Human Melanoma Black- 45; MD: Doctor of Medicine: MelanA Melanoma Antigen; MRI: magnetic resonance imaging; mTOR: mammalian target of rapamycin; NGS: Next generation sequencing; PD: Privatdozent; PeCom: Perivascular epithelioid cell tumors; TFE3: Transcription Factor E3; TSC1: Tuberous sclerosis complex 1; TSC2: Tuberous sclerosis complex 2
Introduction
Angiomyolipoma (AML) is a rare tumour belonging to the family of perivascular epithelioid tumors. Epithelioid angiomyolipoma (EAML) is a variant of the classic angiomyolipoma (AML) [1]. The three typical histopathological components of classic AML, mature fat cells, smooth muscle cells, and (atypical) blood vessels are not present in EAML; instead, epithelioid cells are predominant [2].
EAML accounts for approximately 5-8% of surgically resected AML and shows a tendency towards more aggressive behaviour, which may manifest itself as local recurrence and/or metachronous metastasis to the liver, lymph nodes, or retroperitoneally [3, 4]. It has been assumed in the literature that about 30% of EAML develop malignant potential [5], but this may be an overestimation [1].
Typical AML, because of their benign nature, are usually monitored and treated only when there is a risk of rupture or symptoms, then often by embolization or surgical removal. However, EAML is usually identified only by a histological workup.
Thus, metastatic EAML is very seldom encountered, and there exists no established treatment regimen. Probatory chemotherapies have not yielded satisfactory results [6,7]. However, several case reports and series describe good responses to agents that inhibit the mTOR pathway [8-10].
We here present to our knowledge the first patient in Germany with osseous metastasis of an EAML that showed complete remission after treatment with an mTOR (mammalian target of rapamycin) inhibitor, specifically temsirolimus.
Case Report
We present a 70-year-old male caucasian patient (Pre-therapy: weight 88 kg, height 1.87 m) who had undergone a laparoscopic transperitoneal daVinci-assisted tumour nephrectomy on the left side in 2015 following an incidentally diagnosed mass of the left kidney. Pathological workup of the 8.7 cm large tumour led to the diagnosis of an epithelioid angiomyolipoma (PECom) of the kidney. Histologically, the tumour showed a clear epithelioid morphology with very large voluminous tumour cells and polymorphous enlarged nuclei. Positivity for the melanocyte markers HMB45 and MelanA and negativity for Desmin and S100 were noted. The dignity of the tumour was unclear at the time, with the possibility of a malignant variant due to the size and marked cellular polymorphism (Figure 1). The patient initially presented asymptomatic during follow-up, but a CT scan on 30/09/2019 revealed a lesion in the right os ilium that was highly suspicious of a bone metastasis. Specifically, a 2.5 cm mass of the right os ilium with long corticalis destruction with paraosseous tumour growth was described.
The osseous lesion was confirmed on skeletal scintigraphy performed on 15/10/2019 with additional questionable multi-enhancement at lumbar vertebra 5. In December 2019, a sample was taken from the right ilium after another MRI of the pelvis (Figure 3). Histology revealed a polymorphous tumour cell proliferation with eosinophilic cytoplasm and strong expression of MelanA and HMB45, showing the same immunohistochemical phenotype as the primary lesion. The Actin immunohistochemistry was negative. After further morphological and immunohistochemical consultation and comparative assessment with the primary tumour of the kidney, the finding was confirmed in January 2021 as a metastasis of the epithelioid angiomyolipoma of the kidney (PECom) diagnosed in 2015 (Figure 2).
After this, the patient did not undergo any therapy and initially wanted to obtain a second opinion. When he presented to our institution again in March 2020, the whole-body MRI showed that the os ilium metastasis had progressed to 4.7 cm with further metastases in lumbar vertebra 1 and 3.
Due to the dynamics of the disease, the patient decided to start the mTOR therapy recommended by us with weekly temsirolimus 25mg i.v. The first dose was administered in April 2020. Due to the rapid progression of the metastasis, we initiated the treatment without performing a molecular/ immunohistochemical study of mTOR.
In July 2020, the whole-body MRI showed a very good therapy response of the bone filiae to the baseline of 03.03.2020 with almost completely regressed findings, whereby the intraosseous residue of the Os-ilium metastases showed high ADC values as an indication of cell poverty/viability and the osseous metastases in lumbar vertebra 1 and 3 also showed a therapy response with cell-poor residual findings here in the DWI.
The therapy is tolerated with some well-adjustable side effects. Thus, due to a short-term thrombocytopenia in the 2nd cycle, a therapy break of 1 week was adhered to. Mucositis was treated with mouth rinses. Lower limb oedema improved with oral diuretic therapy with torasemide.
Also in November 2020, there was a very good response to therapy in the os ilium on the right (focal necrosis) and the foci in lumbar vertebra 1 and 3 (constant cell-poor residuals) with no evidence of new tumour metastasis. This maintained for the next months (Figure 4).
To confirm the avitality of the lesions, an FDG-PET/CT was performed on 17/05/2021, which showed no evidence of hypermetabolic local recurrence of the left renal lobe, and in particular showed the known osseous filiae in lumbar vertebra 1 and 3 and in the right os ilium without increased metabolic activity in the sense of avital filiae (Figure 5).
To analyse the underlying biological alterations of the tumour, we ordered a molecular pathological examination. Therefore, a targeted NGS sequencing panel (Oncomine Comprehensive Panel v3, ThermoFisher) was performed, and revealed TSC-2 mutation.
Discussion
AML is a rare mesenchymal tumour usually presenting in middle age (40-50 years) without gender preference. AML is often an incidental finding, but may become symptomatic with flank pain, haematuria or palpable mass and in severe cases hypertension, haemorrhage and renal failure. Treatment options for AML, indicated when symptoms appear, when malignancy is suspected or in asymptomatic cases > 4cm in size, range from active surveillance to embolization to surgical removal via partial resection of the kidney or nephrectomy. Small AML without clinical complications and typical imaging presentation can be monitored because of its usually benign behaviour. However, prophylactically motivated reasons should also be considered, such as women of childbearing age, very large findings and patients in whom follow-up is difficult.
When an AML presents with a low fat component, it can be poorly differentiated from a renal cell carcinoma or retroperitoneal sarcoma by imaging methods, so that a resection should be performed in these cases. Making a definitive diagnosis of EAML by imaging is difficult, partly because the fat content on MRI or CT is often less than 5%, unlike AML. On MRI, it can be helpful to note characteristics such as a T-hypo intensity, tumour necrosis, haemorrhages, cystic changes, exophytic growth, venous invasion or thrombus. In the MR contrast phase, the EAML are usually non-specific. Thus, a definitive diagnosis of EAML can only be made histologically after obtaining tissue by sampling or surgical resection [12,13]. In our patient the MRI showed an osteodestructive lesion with accompanying extraosseous component and strong contrast enhancement with a central recess.
EAML is a rare, but important subtype of AML, since it may follow an aggressive clinical course, which may present as local recurrence or metastasis, preferably in the liver, lung, lymph nodes or retroperitoneum. Tsai et al. report that at least 30% of EAML patients develop a progression to a typical malignant EAML. [17] However, the exact risk of recurrence or metastasis for a given patient is difficult to estimate.
The first descriptions of EAML occurred in the 1990s with individual cases or very small series [14,15]. However, the exact proportion of epithelioid cells that define an EAML is unclear [16]. Various series name <5%, 20% or even 80% as the cut-off value. Enlarged vesicular nuclei and prominent nucleoli are often present. However, atypical nuclei and a high proportion of epithelioid cells may be misdiagnosed as renal cell carcinoma. Therefore, exact workup by an experienced pathologist with additional immunohistochemical tests, such as HMB45 and MelanA, is of great importance for the correct diagnosis of EAML. In our patient the lesion showd a polymorphous tumour cell proliferation with eosinophilic cytoplasm and strong expression of MelanA and HMB45, leading to the diagnosis of malignant EAML.
In addition, histology may provide hints to the clinical course of EAML. For example, Brimo et al. who found ≥ 70% proportion of atypical epithelioid cells, ≥2 nuclear division Figures per 10 major facial fields, atypical nuclear division figures and necrosis. [18]
In 80-90% of the cases, AML is clinically sporadic; in the remaining 10-20% of cases, AML is associated with tuberous sclerosis. [2] . This is an autosomal inherited disease based on inactivating mutations in the TSC1 and TSC2 genes. The proteins encoded by TSC1 and TSC2 are part of a TSC protein complex that negatively regulates mTOR complex 1 and is thus essential for regulating cell growth and size.
Thus, inactivation of TSC1/2 results in overactivation of the mTOR signal transduction pathway. This leads to increased proliferation in the characteristic tuberous sclerosis lesions and tumour development in various organs, including AML in the kidney. However, TSC1/2 alterations are also found in sporadic renal angiomyolipomas [3], and are thought to represent a crucial oncogenic driver. Thus, inhibition of the subsequently activated mTOR signal transduction by drugs represents an important therapeutic option. [11] Since EAML are very rare tumours, no fixed treatment options have been established for EAML. There are cases of complete resection with therapy-free follow-up. Therapeutic approaches with chemotherapies have produced moderate to inadequate results [19,20].
Because our patient had an inoperable lesion as well as 2 other metastases, surgical intervention was not an option. Thus, also in view of the pathogenetic relationship with the mTOR pathway, we chose treatment with an mTOR inhibitor.
Treatment with an mTOR inhibitor, as it also appears reasonable due to the biological basis specifically in cases with TSC1/2 alterations, shows promising results with complete remissions in some cases, as we were able to show in our case [21-23]. However, some cases do not respond to this therapy either, which may be due to a smaller subset of cases harbouring underlying TFE3 fusions [4], and probably additional, not yet revealed pathogenic mechanisms.
In summary, we here present a patient with osseous metastasis of an epithelioid angiomyolipoma with a TSC2 mutation, which showed a complete response to mTOR therapy. This case demonstrates that, although very rare, the correct diagnosis of EAML is important and close follow-up is recommended. Furthermore, treatment with an mTOR inhibitor can succeed in complete remission. Moreover, this case highlights that additional molecular workup may help to guide therapy.
Acknowledgement
The manuscript entitled, “Osseous metastases of renal epithelioid angiomyolipoma with dramatic response through mTOR inhibitor therapy” has not received any funds.
Disclosure of conflict of interest
The authors declare that they have no conflicts of interest.
Author contributions statement
Case report conception and design: Dr. med. S. L. Barton; data collection: Dr. med. S. L. Barton; analysis and interpretation of results: Dr. med. S. L. Barton. Support with pathological findings: Prof. Dr. M. Eck; Dr.med. Dr. rer.nat. E. Gerhard-Hartmann; draft manuscript preparation: Dr. med. S. L. Barton, Dr.med. Dr. rer.nat. E. Gerhard-Hartmann Dr. med. M. Welslau, PD Dr. med. S. Rogenhofer. All authors reviewed the results and approved the final version of the manuscript.
Ethics statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent was obtained from all individual participants involved in the study.
- WCJSP ea He (2013) Epithelioid angiomyolipoma of the kidney: pathological features and clinical outcome in a series of consecutively resected tumors. Mod Pathol 26: 1355-64.
- JWCCJD j BJ Wagner (1997) Adult renal hamartomas. Radiographics : 171-6.
- NHSB Henske EP (1995) Loss of heterozygosity in the tuberous sclerosis (TSC2) region of chromosome band 16p13 occurs in sporadic as well as TSC-associated renal angiomyolipomas,” Genes Chromosomes Cancer: 295-8.
- SYZL Agaram NP (2015) Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications.,” Am J Surg Pathol: 813-25.
- JL Hornick (2006) PEComa: what do we know so far?,” Histopathology: 75-82.
- MRYR Ferry JA (1991) Renal angiomyolipoma with sarcomatous transformation and pulmonary metastases,” Am J Surg Pathol: 1083-8.
- IKNY ea Yokoo H (2000) Retroperitoneal epithelioid angiomyolipoma leading to fatal outcome,” Pathol Int: 649-54.
- DPJCK MAI (1996) Epithelioid cell variant of renal angiomyolipoma. Histopathology 28: 277-80.
- CDIHA Italiano (2010) Treatment with the mTOR inhibitor temsirolimus in patients with malignant PEComa,” Annals of Oncology 21: 1135-7.
- RAMRHY JA Ferry (1991) Renal angiomyolipoma with sarcomatous transformation and pulmonary metastases. Ame J Surgical Pathology: 1083-8.
- JR Sampson (2009) Therapeutic targeting of mTOR in tuberous sclerosis,” Biochemical Society Transactions. 259-64.
- YYAM Kohei Shitara (2011) Dramatic Tumor Response to Everolimus for Malignant Epithelioid Angiomyolipoma,” Japanese Journal of Clinical Oncology: 814-6.
- HM Aydin and CMP Magi-Galluzzi (2009) Renal Angiomyolipoma; Clinicopathologic Study of 194 Cases With Emphasis on the Epithelioid Histology and Tuberous Sclerosis Association,” The American Journal of Surgical Pathology 33: 289-97.
- FM Brimo, BM Robinson, CM Guo (2010) Renal Epithelioid Angiomyolipoma with Atypia: A Series of 40 Cases With Emphasis on Clinicopathologic Prognostic Indicators of Malignancy. The Ame J Surgical Pathol 34: 715-22.
- WKTB Nicholas Wolff (2010) Sirolimus and Temsirolimus for Epithelioid Angiomyolipoma,” JOURNAL OF CLINICAL ONCOLOGY. 28: 65-8.
- MYC, KF N CC Pan (2008) Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): genetic evidence for the relationship of PEComa with angiomyolipoma. J Pathol: 387-93.
- M Chad, H Stone, M Min, W Lee, M Mahul, et al. (2001) Renal Angiomyolipoma: Further Immunophenotypic Characterization of an Expanding Morphologic Spectrum,” Archives of Pathology and Laboratory Med 125: 751-8.
- W J, WC CL (2009) Chia-Chun Tsai, “Epithelioid Angiomyolipoma of the Kidney Mimicking Renal Cell Carcinoma: A Clinicopathologic Analysis of Cases and Literature Review. The Kaohsiung J Med Sci: 133-40.
- BRC G, Fadi Brimo (2010) Renal epithelioid angiomyolipoma with atypia: a series of 40 cases with emphasis on clinicopathologic prognostic indicators of malignancy,” Am J Surg Pathol: 715-22.
- KH L, KF N, Han-Yu Tsai (2019) Clinicopathologic analysis of renal epithelioid angiomyolipoma: Consecutively excised 23 cases,” The Kaohsiung Journal of Medical Sci 33-8.
- KIY N Hideaki Yokoo (2001) Retroperitoneal epithelioid angiomyolipoma leading to fatal outcome,” Pathology International 50: 649-54.
- HM L'Hostis, CM Deminiere, JM M Ferriere (1999) Renal Angiomyolipoma- A Clinicopathologic, Immunohistochemical, and Follow-up Study of 46 Cases,” The American Journal of Surgical Pathology 23: 9: 1011.
- GM Martignoni, MM Pea, FM Bonetti (1998) Carcinomalike Monotypic Epithelioid Angiomyolipoma in Patients Without Evidence of Tuberous Sclerosis. Ame J Surgical Pathol 22: 663-72.
- RKYH NOBUYOSHI TAKAHASHI (2003) Malignant transformation of renal angiomyolipoma. Int J Urology 10: 271-3.
FIGURE 1
Figure 1: Microscopic examination of the nephrectomy specimen revealed an epitheloid tumor with large polygonal cells with eosinophilic cytoplasm and atypical nuclei with prominent nucleoli (HE). The tumor cells showed a positive reaction in the immunohistochemica stainings for HMB45 and MART1 (from left to right), but were negative for AE1/3 as well as PAX8 and SOX10 (not shown). Original magnification 100x, the length of the scale bar is 100�m
FIGURE 2
Figure 2: Microscopic examination of the metastasis specimen. The tumor cells showed a positive reaction in the immunohistochemical stainings for HMB45 and MART1 (from left to right), but were negative for AE1/3 as well as PAX8 and SOX10 (not shown). Original magnification 100x
FIGURE 3
Figure 3: MRI pelvis with Os-ilium metastasis on the right before bioptical confirmation
FIGURE 4
Figure 4: MRI pelvis under temsirolimus therapy 02/2021
FIGURE 5
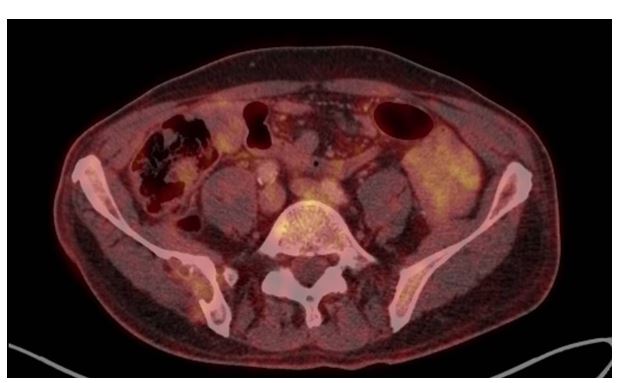
Figure 5: FDG-PET/CT examination 05/2021
Figures at a glance