Basal Ganglia Hyperintensities on MRI, Movement Disorders and High Levels of Manganese in Chronic Hepatic Encephalopathy: A Clinical Study of Cases and Review
Received Date: March 10, 2024 Accepted Date: April 10, 2024 Published Date: April 13, 2024
doi: 10.17303/aglp.2024.1.102
Citation: José M Valera (2024) Basal Ganglia Hyperintensities on MRI, Movement Disorders and High Levels of Manganese in Chronic Hepatic Encephalopathy: A Clinical Study of Cases and Review. J Ann Gastroenterol Liver Pancreatic Dis 1: 1-8
Abstract
Manganese has been associated with chronic hepatic encephalopathy and hyperintensity of the basal ganglia on brain magnetic resonance imaging (MRI).
Aims: to show different clinical presentations of chronic hepatic encephalopathy and MRI abnormalities and their association with blood and cerebrospinal fluid manganese levels, with a review of this entity.
Methods: on 12 patients with neurological symptoms and MRI hyperintensity involving the basal ganglia, blood and cerebrospinal fluid manganese levels were measured, with known or unknown prior chronic liver disease.
Results: patients presented with parkinsonian, choreic or psychiatric symptoms. All patients had cirrhosis, but the diagnosis was known in half of them until MRI was done. High blood levels of manganese was found in all of them (mean:13.2±7.5ug/lt, normal value<0.06) and increased cerebrospinal fluid levels in the 2 cases studied.
Conclusions: Manganese could play a role in the pathogenesis of chronic hepatic encephalopathy on some patients with atypical symptoms. MRI is a useful test of the metabolic brain injury and led to diagnosis of an unsuspected liver disease in some of these patients.
Keywords: Chronic Hepatic Encephalopathy; Manganese Blood Levels; Cerebrospinal Fluid Manganese Levels; Globus Pallidus Hyperintensity; Brain Magnetic Resonance
Introduction
Chronic Hepatic Encephalopathy (CHE) is a disabling complication of chronic liver disease (CLD). The spectrum of clinical manifestations ranges from very mild encephalopathy only detected by careful neuropsychological evaluation [1], to the severe syndrome of acquired hepatocerebral degeneration [2]. The pathogenesis of CHE remains unclear, but increased ammonia levels play a role, although probably not as important as in the acute form of encephalopathy [3,4]. Basal ganglia hyperintensities, mainly affecting the globus pallidus, are a well recognised radiologic finding in magnetic resonance imaging (MRI) of these patients [5,6]. In this sense, a pathogenic role for manganese (Mn) has been implicated in CHE and seems to be the cause of these MRI changes [7-9]. However, to our knowledge, there has been only one report that has documented increased blood and cerebrospinal fluid (CSF) concentrations of Mn in patients with CHE which could lead to its accumulation in the basal ganglia [10]. The aim of this study is to report the clinical features of CHE and the MRI abnormalities found in these patients and their association with elevated Mn levels in blood and CSF, together with a review of what has been published and known about this unusual manifestation so far.
Patients and Methods
We selected adult patients with movement disorders, either attending to the Movement Disorders Clinic of the University of Chile Clinical Hospital with a parkinsonian syndrome, who demonstrated some atypical features such as poor response to levodopa, asterixis, myoclonus and/or gait ataxia, with or without a past history or clinical signs suggesting CLD, as well as patients presenting with chorea of unclear etiology and patients attending the outpatient Gastroenterology Section with a history of CLD who showed a motor disorder on neurological examination. One patient who presented to a psychiatric clinic with mania and a past medical history of CLD was also included. None of the patients reported an occupational exposure to Mn. A clinical evaluation of patients was performed in order to assess the etiology and severity of CLD, including laboratory, radiological and endoscopic tests to establish the Child-Pugh (CP) classification [11] and the presence of portal hypertension (i.e. oesophageal-gastric varices, ascites, splenomegaly). Serum ammonia and its association with Mn levels and CP score were also measured.
The etiology of liver disease was investigated with HBsAg, anti-HBcore, anti-HCV, autoantibodies (antinuclear, anti-smooth muscle and anti-mitochondrial antibodies), immunoglobulin A, G, and M levels, serum ferritin and transferrin saturation, serum caeruloplasmin and the presence of Kayser-Fleischer rings on ophtalmological exam. Ethanol consumption was thoroughly probed during both patient evaluation and interview of the patient’ s family.
Brain MRI images were obtained by a 1.5 Tesla magnet; the magnitude of hyperintensities were not specifically quantified, except for a three grade scale: mild, moderate and severe. After informed consent was obtained, blood levels of Mn were studied in all subjects by atomic absorption spectrometry (Model Varian Spectra A-40). The CSF Mn concentrations were measured in 2 patients who gave informed consent to lumbar puncture (LP), and in whom coagulation studies permitted.
Neuropsychological assessment aimed at detecting frontal lobe dysfunction was performed on patients with CLD unrelated to alcohol (to exclude potential effects of alcohol on the brain) and to those with Wilson’s disease; this evaluation included Mattis Dementia Rating Scale, verbal fluency, Stroop test and Wisconsin Sorting Card Test. Hamilton Rating Scale was used to assess the presence of depression. We used West Heaven scale of liver encephalopathy [1].
The investigation was approved by the local ethics committee.
Results
We studied 12 patients (7 men and 5 women), with a mean age of 56.8 ± 8.6 years, characterized on table 1. The mean duration of neurologic symptoms at the moment of evaluation was 2 years (range: 1- 4 years).
The presenting neurological manifestation was parkinsonism in nine cases, a choreic syndrome in two and a maniac episode in one patient. One of the parkinsonian patients was diagnosed as having Wilson’s disease. Most parkinsonian patients showed symmetric bradykinesia and rigidity with a postural tremor, mild asterixis and gait ataxia. Typical features of Parkinson’s disease with a classic resting tremor was observed in only one patient who showed a poor response to levodopa. The patients who presented with chorea showed symmetric involvement mainly of face and upper limbs associated with dysarthria and gait ataxia. Cognitive impairment suggesting frontal lobe dysfunction was observed in the four cases assessed (patients 1, 3, 6, 9 shown in Table 1), showing poor ability to make executive decisions and verbal fluency, with frequent errors in the Stroop test and in the Wisconsin Sorting Card test. None of the patients were determined to have overt depression.
Only half of the cases had a clear history of CLD preceding the neurological symptoms by a mean period of 6 years (range 4 - 8 years) and were regularly followed by a gastroenterologist. Two cases had experienced episodes of acute encephalopathy, but neither had a history of frank hepatic coma. After a complete evaluation of all patients, the etiology of liver damage was related to alcoholism in six cases, Wilson’s disease in 1 patient, chronic hepatitis C in 1 case, primary biliary cirrhosis in 1 case and cryptogenic cirrhosis in three cases. Liver biopsy had been done in the six patients without alcoholic etiology previously at the time of the study, which confirmed cirrhosis and its etiology in 3 cases (HCV, PBC and Wilson’s disease some years ago) and cryptogenic cirrhosis in the others. In patients with alcoholic consumption cirrhosis was based on the clinical, laboratory and radiological findings.
All patients had highly elevated blood levels of Mn, mean: 13.2 ± 7.5 ug/lt, (normal value < 0.06 ug/lt) [12], and high CSF Mn levels were found in the 2 cases studied, mean 3.5 ± 0.9 ug/dl (normal range: 0.22 - 0.72 ug/dl). Patients with more severe liver damage (CP score B and C) had more elevated Mn blood levels and also showed more severe extrapyramidal features in subjective terms of bradykinesia, rigidity and postural tremor. However, because only one patient with CP score A was included in the study, a significant correlation between Mn blood level, neurological symptoms and severity of CLD could not be determined (Table 1). Mn levels of the 12 patients stratified by CP score are shown in Table 2. Increased arterial ammonia levels were also detected in 10 of the 12 patients.
A characteristic pattern of T1-weighted MRI hyperintensities was found in all cases and involved symmetrically both the globus pallidus and less frequently the caudate nucleus and the midbrain (Figure 1). T2-weighted images were otherwise normal, included the patient with Wilson’s disease. The pattern of basal ganglia hyperintensities was present regardless of the neurological features at presentation, and tended to be more intense in those patients who presented with more severe neurological symptoms (Table 1).
A mild and intermittent clinical improvement was seen after medical treatment of the encephalopathy with lactulose and non-absorbable antibiotics in some patients with parkinsonian symptoms in terms of gait, hand tremor and verbal fluency, those who had baseline levels of blood Mn and serum ammonium somewhat higher initially (patients 2, 5, 6, 7, 10 in Table 1) without becoming a relationship statistically associated with the magnitude or etiology of CLD.
Discussion
Our report confirms the presence of basal ganglia hyperintensity on T1-weighted MRI images in patients with CHE independent of the underlying etiology of the CLD [13-15]. This finding has been described in approximately 90% of cirrhotic patients [9] even in the absence of overt encephalopathy, but they have tended to be more severe in the more advanced cases, such as those patients with CHE [7,13]. These MRI hyperintensities along with the neurologic impairment may be completely reversible after successful treatment of the hepatic disease by liver transplant [16,17], which none of our patients had received as of the writing of this manuscript.
We found elevated blood Mn in the cirrhotic patients in this cohort irrespective of the etiology. Of the 12 patients included in our study, only half had known CLD and the others presented with neurological features, i.e. extrapyramidal manifestations, which prompted MRI study and investigation for CLD. Actually, the MRI findings in a subgroup of patients with these neurological symptoms may suggest that evaluation for occult liver damage is warranted. There was only one case with CP score A and minimal changes on imaging. This suggests that studying other patients with mild liver disease may better define the relationship between the severity of CLD, MRI findings and Mn levels.
Increased Mn blood levels were found in all cases, and in the CSF of the two cases studied with LP. These findings suggest a role for Mn deposition in the pathogenesis of CHE. However, controversy still exists about the role of Mn in CHE, since some reports show an elevation of Mn blood levels in almost 70% of cirrhotics [9], whereas other authors have not been able to detect such an association [18]. It is difficult to explain this discrepancy, although as is true with ammonia determinations, Mn levels must be evaluated frecuently to detect an elevation which may not be permanent. However, in some cases like the one reported by Thobois et al. [18], normal Mn blood levels were found throughout the follow up, and more recently Maffeo, et al. [19] in 26 patients with cirrhosis and T1 hyperintensity (only seven with CHE) did not find that blood levels of Mn reflect the MRI signal or CHE and concluded that MRI signal is a prerequisite for clinical CHE. With the possible fluctuations in blood Mn levels, CSF Mn determination may be a better marker of Mn brain deposition than blood levels, as suggested by Katsuragi, et al. [10]. We found elevated blood levels of Mn in all our patients, but with movement disorders being an inclusion criteria in our cohort, so this finding may be explained by selection bias. Furthermore, it´s interesting to evaluate an additional group of patients with CLD without neurological improvement to correlate with Mn levels, MRI imaging and evolutive neurological symptoms in all of them, as the study mentioned above suggests [19].
Direct evidence of Mn basal ganglia deposition has been provided simultaneously by Krieger et al (7) and Pomier-Layrargues, et al. [20], who detected increased Mn tissue levels in the basal ganglia at neuropathological examination of cirrhotic patients in comparison to a control group. The characteristic hyperintensity on brain MRI have also been observed in patients with an incorrectly formulated total parenteral nutrition solution with an excess of Mn supplementation [21]; these abnormal findings disappeared after correction of the Mn prescribed [21,22]. Huang et al demonstrated the same MRI hyperintensity in humans with an occupational exposure to Mn [23]. In all of these observations, as in our report, the pattern of basal ganglia hyperintensities is constant with a clear predilection for the globus pallidus, caudate nucleus, midbrain, and less frequently, the white matter. This pattern of lesion distribution is unrelated to the presenting movement disorders since we observed a similar distribution of lesions whether the presentation was parkinsonian, choreic or psychiatric. This fact could suggest that these images constitute only a secondary finding, such as an unspecific response of some more vulnerable areas of the brain as Jog and Lang suggested in their review [13]. However, in our opinion, the constant distribution of lesions as seen on MRI in these patients would not necessarily correspond with clinical presentation. Furthermore, the symmetric clinical findings in all patients of our series correlate with a symmetric radiological involvement of the basal ganglia nuclei. It is noteworthy that one of our patients presented initially with prominent psychiatric manifestations of a maniac episode with impulsivity, hallucinations and delusions. These symptoms resembled what has been referred to as manganese mania or manganese psychosis, documented mainly in manganese miners [24], suggesting the importance of Mn levels and imaging changes in this subgroup of CLD patients.
Mn is normally excreted by the liver into the biliary system, thus in the presence of liver damage and/or porto-systemic shunt in acute and chronic cases, an increase in their levels and those of ammonia is explained, whose metabolic enzyme glutamine synthetase uses Mn as a cofactor, which would also contribute to his accumulate in the brain with a favorite dilection for the basal ganglia [3]. Both ammonia and Mn specifically bind to a benzodiazepine receptor called peripheral-type benzodiazepine receptor (PTBR) that is located mainly on the outer mitochondrial membranes of astrocytes. It has been suggested that activation of PTBR in the brain by exposure to ammonia or manganese potentiates inhibitory gabaergic neurotransmission which is characteristic of CHE (4). Within neurons, Mn is sequestered within mitochondria where it can impede oxidative phosphorylation and decreases the activity of mitochondrial enzymes [25,26]. Mn in vitro has also been demonstrated to stimulate nitric oxide synthetase activity, leading to an increase of this potential neurotoxin [26]. Some evidence also suggests that Mn could affect dopaminergic transmission [25].
In recent years, great emphasis has been placed on the concept of subclinical hepatic encephalopathy, or as it is more properly called, mild or minimal encephalopathy [1], which includes mild psychological deficits in the absence of overt encephalopathy. These deficits which clearly affect patient daily life include difficulties in decision-making, attention, and verbal fluency, as was seen in some of our patients. We hypothesize that basal ganglia lesions seen on brain MRI could explain the frontal lobe deficit underlying the encephalopathy of CLD patients as seen in other basal ganglia disorders like Parkinson’s disease, although that is usually seen later in this condition. In addition, in CHE its management is more limited with poor clinical response as described with the use of dopaminergic agents [27,28], non-absorbable disaccharides or classic copper chelators as described in a Wilson-like case of a 55-year-old man [29]. Some improvement has been reported in isolated cases with the use of agents that may lag Mn, such as Trientene, in which T1 hyperintensity and clinical parkinsonism decreased in a patient [30] or neurological engagement was at least stabilized in the case mentioned above [29]. Better clinical and radiological outcome has been reported with obliteration of portal-systemic shunt in CHE [31], confirming the importance of this matter in the pathophysiology of this alteration. This image has also been described in T1 brain MRI in six patients with portal venous thrombosis-induced shunt with cavernomatosis, with no signs of liver disease or portal-systemic encephalopathy [32]. There are few cases described after the therapy more definitive which means the liver transplant, communicating partial to complete clinical resolution or satisfactory on images [17,19,33-35], as well as of the post-transplant transitional improvement and subsequent recurrence associated to graft dysfunction [36], and strangely the persistence of mild parkinsonism in patients with minimal pre-transplant liver encephalopathy and hyperintense signal in MRI palidus [37].
Of course, we must recognize the limitations of our study in terms of the small number of patients and the lack of a control group with CLD without movement disorders in which we observed basal ganglia on MRI and Mn blood levels. However, in this infrequent area of atypical presentation of CHE it seems important to us to describe this type of cases that requires specific study, search for treatment and follow-up with and without liver transplantation.
In summary, we believe that our report adds to the hypothesis that Mn plays a role in the pathogenesis of CHE and that brain MRI is an important test that suggest metabolic brain injury in this context. Strategies to avoid Mn deposition in the brain, not yet available or obliteration of shunts in severe cases, could be an option to treat the encephalopathy and atypical neurological symptoms of these patients before liver transplantation, or when such treatment is unfeasible. To better understand the pathogenic role of Mn in CLD, further perspective research is necessary to correlate blood and CSF Mn levels. MRI findings, neurologoical symptoms and survival differnces among patients with mild to severe CLD.
- Lockwood A (2002) Hepatic Encephalopathy. In: Albers J, Berent S. Editors. Neurologic Manifestations of Systemic Disease. Neurologic Clinics, Philadelphia Saunders 20: 241-6.
- Victor M, Adams R, Cole M (1965) The acquired type of chronic hepatocerebral degeneration. Med 44: 345-96.
- Albretch J, Jones E (1999) Hepatic Encephalopathy: Molecular mechanisms underlying the clinical syndrome. J Neurol Sci 170: 138-46.
- Butterworth R (2000) Hepatic Encephalopathy: a neuropsychiatric disorder involving multiple neurotransmitter systems. Curr Opin Neurol 13: 721-7.
- Kulisevsky J, Ruscalleda J, Grau JM (1991) MR imaging of acquired hepatocerebral degeneration. AJNR 12: 527-8.
- Inoue F, Hori S, Narumi I (1991) Portal systemic encephalopathy: presence of basal ganglia lesions with high signal intensity on MR images. Radiology 179: 551-5.
- Krieger D, Krieger S, Jansen O, Gass P, Theilman L, et al. (1995) Manganese and chronic hepatic encephalopathy. Lancet 346: 270-4.
- Hauser R, Zesiewicz T, Rosemurgy A, Martinez C, Olanow C (1994) Manganese intoxication and chronic liver failure. Ann Neurol 36: 871-5.
- Spahr L, Butterworth RF, Fontaine S, Bui L, Therrien G, et al. (1996) Increased blood manganese in cirrhotic patients: relationship to pallidal magnetic resonance signal hyperintensities and neurological symptoms. Hepatology 24: 1116-20.
- Katsuragi T, Iseki E, Kosaka K, Koyano S, Twabuchi K. Cerebrospinal fluid manganese concentrations in patients with symmetric pallidal hyperintensities on T1 weighted MRI. J Neurol Neurosurg Psychiatry 2000: 531-2.
- Infante-Rivard C, Esnaola S, Villeneuve J (1987) Clinical and statistical validity of conventional prognostic factors in predicting short-term survival among cirrhotics. Hepatology 7: 660-4.
- Falchuk K (1998) Disturbances in trace element metabolism. In: Isselbacher K, Braunwald E, Wilson J, editors. Harrison’s Principles of Internal Medicine, 14th ed. New York: McGraw-Hill 1998: 557-8.
- Jog M, Lang A (1995) Chronic Acquired Hepatocerebral Degeneration: Case reports and new Insights. Mov Disord 10: 714-22.
- Pujol A, Pujol J, Graus F, Rimola A, Peri J, et al. (1993) Hyperintense globus pallidus on T1-weighted MRI in cirrhotic patients is associated with severity of liver failure. Neurology 1993: 4365-9.
- Saito H, Ejima A (1995) Liver dysfunction and probable manganese accumulation in the brainstem and basal ganglia. J Neurol Neurosurg Psychiatry 58: 760-1.
- Powell E, Pender M, Chalk J, Parkin P, Strong R, et al. (1990) Improvement in Chronic Hepatocerebral Degeneration Following Liver transplantation. Gastroenterology 98: 1079-82.
- Stracciari A, Guarino M, Pazzaglia P, Marchesini G, Pisi P (2001) Acquired Hepatocerebral degeneration: full recovery after liver transplantation. J Neurol Neurosurg Psychiatry 70: 136-7.
- Thobois S, Giraud P, Debat P, Gouttard M, Maurizi A, et al. (2002) Orofacial Dyskinesias in a Patient with Primary Biliary Cirrhosis: a clinicopathological case report and review. Mov Disord 17: 415-8.
- Maffeo E, Montuschi A, Stura G, Giordana MT (2014) Chronic acquired hepatocerebral degeneration, pallidal T1 MRI hyperintensity and manganese in a series of cirrhotic patients. Neurol Sci 35: 523-30.
- Pomier-Layrargues G, Spahr L, Butterworth R (1995) Increased manganese concentrations in pallidum of cirrhotics patients. Lancet 345: 735.
- Mirowitz S, Westrich T, Hirsch J (1991) Hyperintense basal ganglia on T1-weighted MR images in patients receiving parenteral nutrition. Radiology 181: 117-20.
- Mirowitz S, Westrich T (1992) Basal ganglia signal intensity alterations: reversal after discontinuation of parenteral manganese administration. Radiology 185: 535-36.
- Huang CC, Chu NS, Lu CS, Wang JD, Tsai JL, et al. (1989) Chronic manganese intoxication. Arch Neurol 46: 1104-6.
- Trimble M, Krishnamoorthy E (2000) The role of Toxins in Disorders of Mood and Affect. In: Albers J, Berent S, editors. Clinical Neurobehavioral Toxicology. Neurologic Clinics. Philadelphia Saunders 18: 649-63.
- Albin R (2000) Basal Ganglia Neurotoxins. In: Albers J, Berent S, editors. Clinical Neurobehavioral Toxicology. Neurologic Clinics. Philadelphia Saunders18: 665-80.
- Brouillet E, ShinolbuL, Mc Garvey U, Hochberg F,Beal M (1993) Manganese injection into rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp Neurol 120: 89-94.
- Tuschl K, Mills PB, Parsons H (2008) Hepatic cirrhosis, dystonia, polycythaemia, and hypermanganesaemia—a new metabolic disorder. J Inherit Metab Dis 31: 151–63.
- Butterworth RF (2013) Parkinsonism in cirrhosis:pathogenesis and current therapeutic options: Metab Brain Dis 28: 261-7.
- Rajoriya N, Brahmania M, J Feld J (2019) Implications of Manganese in Chronic Acquired Hepatocerebral Degeneration. Ann Hepatol 18: 274-8.
- Park HK, Kim SM, Choi CG, Lee MC, Chung SJ (2008) Effect of trientine on manganese intoxication in a patient with acquired hepatocerebral degeneration. Mov Disord 23: 768-70.
- Hisahara S, Matsushita T, Kitamura M, Mezawa S, Nonaka M, et al. (2014) Long-term clinical and radiological improvement of chronic acquired hepatocerebral degeneration after obliteration of portosystemic shunt: Report of a case. J Neurol Sci 346: 303-6.
- Nolte W, Wiltfang J, Schindler CG, Unterberg K, Finkenstaedt M, et al. (1998) Bright basal ganglia in T1-weighted magnetic resonance images are frequent in patients with portal vein thrombosis without liver cirrhosis and not suggestive of hepatic encephalopathy. J Hepatol 29: 443-9.
- Stracciari A, Baldin E, Cretella L, Delaj L, D'Alessandro R, et al. (2011) Chronic acquired hepatocerebral degeneration: effects of liver transplantation on neurological manifestations. Neurol Sci 32: 411-5.
- Pinarbasi B, Kaymakoglu S, Matur Z, Akyuz F, Demir K, et al. (2009) Are acquired hepatocerebral degeneration and hepatic myelopathy reversible? J Clin Gastroenterol 43: 176-81.
- Pigoni A, Iuculano F, Saetti C, Airaghi L, Burdick L, et al. (2018) Acquired hepatocerebral degeneration (AHD): a peculiar neurological impairment in advanced chronic liver disease. Metab Brain Dis 33: 347-52.
- Servin-Abad L, Tzakis A, Schiff ER, Regev A (2006) Acquired hepatocerebral degeneration in a patient with HCV cirrhosis: complete resolution with subsequent recurrence after liver transplantation. Liver Transpl 12: 1161-5.
- Lazeyras F, Spahr L, DuPasquier R, Delavelle J, et al. (2002) Persistence of mild parkinsonism 4 months after liver transplantation in patients with preoperative minimal hepatic encephalopathy: a study on neuroradiological and blood manganese changes. Transpl Int 15: 188-95.
FIGURE 1
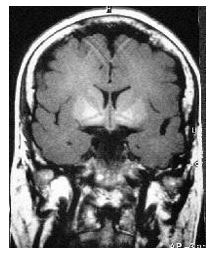
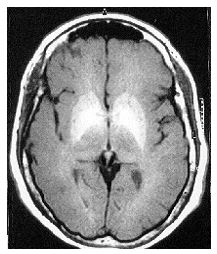
Figure 1: a) Axial, and b) Coronal T1-weighted magnetic resonance imaging of the brain showing basal ganglia hyperintensities, more marked in globus pallidus, caudate nucleus and mesencephalon in a patient with chronic hepatic encephalopathy
Tables at a glance
Figures at a glance