Microbe Mysteries and DNA Dazzles: Navigating Diagnoses and Crime Scenes with Next-Gen Sequencing
Received Date: November 15, 2024 Accepted Date: December 15, 2024 Published Date: December 18, 2024
doi: 10.17303/jbb.2024.2.102
Citation: Aayushi Sanwal, Dr. Umema Mohsin (2024) Microbe Mysteries and DNA Dazzles: Navigating Diagnoses and Crime Scenes with Next-Gen Sequencing. J Biotechnol Biol 2: 1-12
Abstract
The first next-generation sequencing (NGS) technologies appeared approximately ten years ago, which has drastically altered the methods used in genetic research. Complete genomes are now mapped and released nearly every week at a rate that is both faster and less expensive than before. The last ten years have seen the development of NGS platforms and methodologies, and sequence quality has increased to the point that NGS is being utilized for human clinical diagnosis. NGS technologies have also been investigated by forensic genetic laboratories, and in the past year in particular, there has been a little surge in the number of research publications and conference presentations addressing the forensic applications of NGS. This review has focused on the NGS in forensic genetics and its application in clinical microbiological cases.
Keywords: NGS; Forensic STR; Bioinformatics; Bacterial Imprint
Introduction
The first technique for sequencing DNA was published over twenty-five years after the structure of DNA was found. This technique included the insertion of chain-terminating dideoxynucleotides that were either fluorescently or radioactively tagged (previous approach) to accomplish the sequencing of a DNA strand complementary to the probed template strand [1]. The sequencing of the fragments was then ascertained by size separation and gel electrophoresis analysis. The technique, called Sanger-sequencing, was widely adopted as a "first-generation sequencing" technique for sequencing small and big genomes, ranging from those of bacteria and phages to humans [2]. It improved further with the advent of capillary electrophoresis. The method's throughput was limited since it could only assess one sequencing reaction at a time. Next-generation Sequencing (NGS) was introduced between the years 2004 and 2006 and transformed the biomedical discoveries that resulted in an enormous rise in sequencing the data output [3]. The concepts and advancements in nanotechnology that made it possible to sequence individual DNA molecules in massively parallel were responsible for the notable rise in data production. NGS is distinguished by its unique combination of high throughput and single-molecule DNA sequencing capabilities, regardless of the sequencing platform. Over the past several decades, there has been an upsurge in the use of next-generation sequencing (NGS) technology in clinical diagnosis [4]. Three distinct uses of current NGS technology may be found in clinical microbiology laboratories: shotgun metagenomics sequencing, targeted metagenomics sequencing, and whole genome sequencing (WGS) [5].
Forensic genetics has a lengthy history with DNA sequencing. Years before the first short tandem repeat (STR) assays were created, in the late 1980s and early 1990s; sequencing of mitochondrial DNA (mtDNA) was assessed and used for casework. At the time, restriction fragment length polymorphism (RFLP) investigation was the latest state of the field for human beings [6]. The European DNA Profiling (EDNAP) Group's mitochondrial DNA population database project (EMPOP) was started in 1999 to develop an online mtDNA database containing high-quality mtDNA population data and a common forensic standard for mtDNA sequencing [7].
Sequencing was accomplished by the Sanger dideoxynucleotide (ddNTP) chain termination technique, where the insertion of a ddNTP to a developing DNA chain halted further extension by the DNA polymerase. Complete genome sequencing became feasible with the advent of fluorescently labelled ddNTPs and capillary electrophoresis (CE) technologies, which improved throughput and sensitivity while bringing down the cost of Sanger sequencing [8].
According to [9] in 1996, real-time sequencing alternatives to Sanger sequencing emerged, notably pyrosequencing. This method utilized the release of pyrophosphate during DNA synthesis to drive a cascade of enzymatic reactions involving ATP Sulfurylase, Luciferase, and Apyrase, producing detectable light in real-time. By eliminating the need for electrophoresis, pyrosequencing offered a faster and more cost-effective alternative to traditional methods. Despite these advantages, its short sequencing lengths and limited multiplexing capabilities posed challenges, particularly for analyzing the small DNA quantities typical of trace samples [10].
Although pyrosequencing itself did not achieve widespread adoption, it introduced critical innovations—real-time observation of sequencing events and enzymatic signal generation—that underpinned the development of NGS technologies. The first high-throughput sequencer, the Genome Sequencer 20 from 454 Life Sciences, applied pyrosequencing to sequence the human genome within five months at a fraction of the cost of the Sanger method [11]. This breakthrough marked a pivotal shift towards high-throughput sequencing systems that could generate massive datasets more efficiently. Subsequent technologies, such as Solexa (later acquired and renamed Illumina), leveraged these foundational principles to create scalable platforms capable of handling diverse applications, from whole-genome sequencing to transcriptomics and metagenomics. These advancements transformed DNA sequencing from a labor-intensive task into an accessible, high-speed tool that drives modern research and clinical diagnostics [12].
Evolution of Sequencing Technologies
In the past 60 years, sequencing technologies have expanded quickly, beginning with first-generation sequencing and progressing to third-generation sequencing technologies at present. First-generation sequencing began at the same time that Frederick Sanger and the collaborative team of Allan Maxam and Walter Gilbert revealed their procedures for sequencing DNA [13]. When scientists assessed pyrophosphate production using luminescence to ascertain the nucleotide sequence rather than using radiolabeled or fluorescently labeled nucleotides, the second generation of sequencing technologies was born. A large parallel of sequence reactions could occur because of the technique known as "pyrosequencing" and the equipment designed to carry it out increases the amount of DNA that could be sequenced in a single run. Additional techniques for sequencing in parallel developed over time [14]. Each solitary original flow-cell binding DNA strand is bound; a solid phase polymerase chain reaction (PCR) creates clusters of clonal populations from it. Compared to previous second-generation methodologies, this paired-end data gives more information, better accuracy, and lower background noise [15].
Nowadays, the advancement of second- and third-generation sequencing has significantly transformed both clinical microbiology and forensic genetics. In clinical settings, the shift from traditional culture-dependent methods to metagenomics sequencing has allowed to make the rapid and comprehensive pathogen detection, even in polymicrobial infections or low-biomass samples. For example, whole genome sequencing (WGS) has been pivotal in tracking antibiotic resistance genes and understanding bacterial phylogenetics in outbreak scenarios.
In forensic genetics, advancements in sequencing technologies have enhanced the ability to analyse degraded DNA samples, which was previously unattainable with first-generation methods. Techniques such as sequencing-by-synthesis have been employed to reconstruct genetic profiles, aiding in solving long-standing criminal cases and establishing familial relationships.
Microbial Whole Genome Sequencing
The method of sequencing and assembling a microbiological organism's genome of interest is known as whole genome sequencing or WGS. For organisms that are hard to grow or cannot develop in culture, this is a constraint. WGS is used for viral genomes by directly sequencing the material to identify the desired viral genome [16].
WGS may be used to identify an organism, type an organism for epidemiological purposes, and find potential antibiotic resistance in an organism by culturing and isolating the microbe. The first steps in the identification of cultured bacteria can be identified using standard clinical microbiology techniques such as basic morphological observations, biochemical tests, and matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF MS), which is still much faster and more accurate than WGS. Although MALDI-TOF MS can now distinguish between these two Enterococcus species rather well, this is a severe illustration of why genus identification is not always enough [17]. The extremely common ST-452 lineage, which is associated with clonal complex CC23 and has been reported from many nations, is of interest. The initial event and founder strain of ST-452 may have resulted from a single gene transfer between the hyper virulent CC17 lineage and CC23, according to WGS and phylogenetic analysis of the core genome [18]. In certain instances, more fastidious pathogens, like Mycobacterium chimaera, cause epidemics in hospital settings. In these scenarios, WGS is utilized for both identification and epidemiologic tracking of the picky organisms. After these M. chimaera isolates were sequenced, it was discovered that there was a genetic and epidemiological connection between the infections and the water heater-cooler units that these patients had undergone cardiac operations [18].
Metagenomics Sequencing from direct Sample
One significant benefit of using metagenomics sequencing straight from a clinical sample when considering the future of next-generation sequencing (NGS) in clinical microbiology is the removal of the need for culture. This can significantly speed up response times and give medical professionals answers more quickly. To identify pathogens directly from a clinical sample, two distinct NGS metagenomics sequencing techniques may be applied.
First, certain pathogen primers are added to the extracted DNA to amplify the desired group of pathogens (such as bacteria or fungi) or a single pathogen of interest (such as HIV, or SARS-CoV-2). This enrichment method is called deep amplicon or focused sequencing.
Targeted Sequencing
Targeted sequencing is a sequencing technique where an organism or a subset of organisms of interest are subjected to a selection or enrichment procedure, either before or after the library creation step (Figure 1). The benefits of all these techniques include less interference from human DNA and increased sensitivity of detection in sample types containing high concentrations of human cells (e.g., tissue or sputum) [47]. The number of infections that can only be detected using these focused sequencing technologies is its main drawback. Both second and third-generation technology platforms have been used to carry out targeted sequencing straight from samples [19].
Deep amplicon sequencing is another name for PCR amplification in targeted sequencing. Advancement in PCR technique that provides deeper coverage of the targeted gene or genes is deep amplicon sequencing [20]. For deep amplicon sequencing with both bacterial and fungus identification, several labs have verified and deployed LDTs; one group even demonstrated a helpful process to include both. Furthermore, the utilization of 16S deep amplicon sequencing has facilitated the identification of more challenging species, such as bacteria carried by ticks, which are often undetectable by traditional bacterial culture [21].
Shotgun Metagenomics Sequencing
Shotgun metagenome sequencing is a technique that, in contrast to targeted sequencing, casts a larger net since every nucleic acid in a sample is sequenced (Figure 1). Almost all pathogens, including bacteria, fungi, viruses, and parasites, can be detected with a single test thanks to whole genome sequencing. Sequencing has been shown effective in identifying infections from a wide range of material types, including typically sterile sources like joint fluid and CSF blood [23]. Furthermore, it has been shown to assist in the identification of an infectious agent in specimen categories, such as respiratory tract specimens, when a documented microbiome is present. Urine and gastrointestinal specimens. This methodology has been applied as an LDT in several laboratories, which function as patient clinical reference testing centres [24]. The first is a comprehensive metagenomics sequencing test of CSF offered by the University of California, San Francisco (UCSF) clinical microbiology laboratory, which is CLIA-certified. With a clinical sensitivity and specificity of 73% and 99%, respectively, this test has an overall accuracy of 90%. Numerous case reports, including those involving Taenia soliuminfection, West Nile virus, and neuro brucellosis, demonstrate the diagnostic value of this test in situations where all other conventional diagnostic testing was negative. Compared to the traditional direct detection testing of CSF (culture, antigen testing, or quick molecular approaches), the shotgun metagenomics CSF test detected a higher number of infections [25].
The Fundamentals of NGS: Forensic Insights
Some NGS systems are so powerful that the sequencing assay's goal could be as simple as sequencing every double-stranded DNA molecule present in the sample. Shotgun sequencing sometimes referred to as RNA sequencing or RNAseq, may provide an RNA sequencing or RNAseq gene expression profile if the sample material is cDNA [26].
The alternative to shotgun sequencing is commonly referred to as targeted (re-) sequencing. It starts with an enrichment stage in which the selected sections are either amplified by PCR, captured using probes, or captured using a mix of enzymatic processes and probes. The probes can either be biotinylated and combined with their targets in solution, or they can be affixed to a solid surface (like a slide or a bead). In either case, the goal is to remove the undesirable DNA fragments and capture the chosen genomic areas. After that, the DNA is extracted and put to use in sequencing [27,28]. The businesses listed above also provide services for creating personalized panels that the customer specifies for certain tasks or objectives. The main benefit of capture approaches is that the areas of interest receive the majority of the sequencing capacity. While probe-based approaches usually require 50–500 ng of DNA per multiplex reaction, the PCR-based capture method is among the most sensitive and only needs <10 ng. On the other hand, the number of multiplexing capabilities limits PCR-based catches (with the Ion AmpliSeqTM technology, up to 6144 amplicons) [48]. The DNA fragments are utilized to create a library after it has been ready for capture or shotgun sequencing. One or two PCR reactions in which the PCR primers are labelled with sequences required for the subsequent reactions are used to build the library, or adapters can be ligated to the fragments. Certain sequences for the clonal amplification of the library, target sequences for the NGS reaction, a key sequence of 4–8 nucleotides are used for NGS reaction quality control, and a 6–10 nucleotide barcode for sample identification may be included in the adapters or PCR primer tags [29].
NGS in Forensics Genetics
A forensic geneticist is fascinated by the prospect of sequencing every DNA molecule present in the sample. These scientists are accustomed to overcoming the difficulties of extracting meaningful information from trace samples, which frequently comprise DNA from many contributors. Shotgun sequencing, however, is inapplicable for much forensic evidence and necessitates micrograms of DNA [30]. Furthermore, since the shotgun experiment generates sequences from random locations in the genome rather than targeting specific targets, replication could be hard. As a result, two shotgun sequencing trials using the same sample, for example, a reference sample from a suspect and a trace sample will produce different outcomes. Studies comparing platforms have shown that 20% of SNPs and 80% of insertions/deletions (indels) called by one platform could not be replicated by typing the identical material on another [31]. A significant amount of the discrepancies was due to systematic mistakes brought about by the various alignment and variant calling techniques, or they were seen in areas with little to no sequencing depth for one or both platforms. Some NGS systems have enormous capacities, yet they can barely sequence genomes larger than the size of the genome of a human being, and in most shotgun sequencing operations, only a small fraction of the genome is covered by many reads [32].
Ultimately, in the majority of forensic genetic cases, where the main goal is to determine the identity of the individual or individuals contributing to the sample and potentially estimate any phenotypical characteristics of the individual(s) or identify the specific tissue type(s) in the sample, whole genome sequencing seems excessive. This calls for comparatively few markers, therefore a capture-based strategy will use less sample material and be far more cost-effective. These days, PCR-CE is used to type the key forensic markers. Individual tests are available for autosomal, Y-chromosome, X-chromosome, indels, mtDNA, autosomal, and Y-chromosome SNPs, as well as phenotypical, mRNA, ancestry informative markers (AIMs), and other forensic markers [33]. While NGS requires at least two to three days, PCR-CE may be completed in a single workday. However, if a capture for the pertinent loci can be developed, then one of the main benefits of NGS is the ability to integrate all or most of the PCRCE tests into a single NGS assay.
However, creating a combined multiplex PCR will be extremely challenging due to the wide diversity in target copy counts [34]. It's crucial to remember that genomic DNA should not be included in cDNA analysis and that semi-quantitative mRNA sequencing is required for sample tissue identification. Nevertheless, before building the library (using adaptor ligation or the second PCR), it could be able to combine PCR results from distinct PCR captures of nuclear DNA, mtDNA, and cDNA. Or, more frequently, to combine libraries of cDNA, mtDNA, and DNA before clonal amplification. In this manner, all pertinent markers ought to be able to be sequenced in a single sequencing process.The information gleaned from mRNA and mtDNA is not always required, and the case officer must process all sequencing data if it is generated, even if the data is unrelated to the case, so it is unclear if this is a workable solution for casework [35. Therefore, it will be preferable to use versatile NGS systems for a variety of scenarios, including assays with a large number of markers [36].
STR Sequencing
Because there are sizable national DNA databases containing STR profiles from criminal offenders and priceless trace samples from past cases, STRs are and will remain crucial to crime casework. As a result, the essential STR loci need to be able to be sequenced by any NGS assay created for forensic genetics. Though repeats make up about half of the human genome and STRs alone 15%, repeats have not received as much attention as SNPs, minor indels, and copy number changes in NGS research [36]. The majority of instruments' read lengths were too short to cover many repeat structures in the years after the introduction of the first NGS platforms, making it challenging to align reads containing repeating sequences and frequently leading to their disregard. The majority of forensic literature using NGS STR data was created using pyrosequencing technology as these platforms were the only ones with long enough read lengths to sequence the fundamental STR loci utilized in forensic genetics. Sequencing via synthesis and, more recently, semiconductor sequencing was employed in a few publications. Direct PCR or adapter ligation was used to create the libraries. The real variance of STR loci is revealed by sequencing, as opposed to fragment length analysis using PCR-CE. While sequencing of simple STRs has shown few novel alleles, NGS of mostly complex and compound STRs has revealed previously unidentified STR alleles as well as increased overall variability [37]. Additionally, sequence variants can be discovered in the areas around STRs, such as the rs6736691 SNP next to D2S1338. Two families of SNP-STR haplotypes and a two-fold increase in the number of SNP-STR alleles can be produced by a single SNP with a minor allele frequency of a suitable size [37].
Commercial kits of NGS for forensic genetics
The HID-Ion AmpliSeq TM Identity Panel, which amplifies 124 autosomal SNPs, including the majority of SNP for ID and Individual Identification SNPs (IISNPs), as well as 34 Y-chromosome SNPs, was introduced by Thermo Fisher Scientific in 2014 and is intended for use with the Ion PGM TM System. Additionally, the HID-Ion AmpliSeq TM Ancestry Panel, which includes the majority of the Ancestry Informational indicators (AIMs) in the Seldin [38] and Kidd laboratory selection panels [39], is intended for use in ancestry estimation.
Creating a forensic NGS tool for sequence data processing and reporting will be one of the main difficulties. It is not feasible to manually evaluate the sequences or even each genotype call with NGS data. Therefore, before they can be utilized in actual casework, the software solution needs to be totally reliable and evaluated fully [45]. Software tools for kit analysis have been developed by Thermo Fisher Scientific and Illumina1, but they are not advanced enough for forensic genetics. Distinct laboratories have traditionally specified distinct analysis parameters based on internal validation studies and different accreditation requirements [40]. It is widely known that different loci need different criteria for analysis. Estimating bio-geographic ancestry, mtDNA haplogroups, Y-chromosome haplogroups, tissue identification, and phenotypes should also be done using future software modules. Additionally, the software algorithms must be thoroughly explained in the user manual or scientific publications. The assessments need to apply to the ISFGs and other comparable forensic standardizing groups' recommendations [41].
New aim of NGS in forensic genetics
NGS allows forensic genetic studies to be expanded into new forensic medicine-related domains in addition to the examination of traditional forensic markers. Shotgun or exome sequencing may be able to uncover genetic variations linked to recognized illnesses in cases of unexpected and unexplained death, helping the pathologist determine the cause of death. Sudden unexpected cardiac arrest is thought to be the cause of 20% of deaths in Denmark, and even after an autopsy, one-third of these cases are still unsolved. Many of these people are thought to have a genetic illness, and variations linked to the disease could be found through the sequencing of certain genes [42]. Genetic testing can help identify relatives who share a genetic problem and start treating them, in addition to increasing the diagnosis rate and providing valuable information for research on heart disorders. Similarly, NGS may be employed to screen for variations in genes implicated in specific drug metabolism and to support toxicology examinations of a deceased individual to determine if an unexpected death was intentional or unintentional. Additionally, it will be feasible to examine the DNA of the deceased's bacteria, viruses, phages, and fungus to either identify the microorganisms that cause the disease or search for imbalances in the microbial communities that can provide information about the cause of death [43. Furthermore, it was shown that just 50% of the microbiome diversity was shared by samples that were obtained simultaneously, a few meters away. However, there was a lot more variety between the sample locations [44].
Challenges and Advancements in Clinical NGS and Human Identification
Next-generation sequencing (NGS) has revolutionized molecular diagnostics, yet we face many challenges, especially in the context of mixed infections or rare pathogens in clinical environment. Mixed infections often involve multiple pathogens that might vary and are widely in abundance and genetic similarity. In such cases, the interpretation of sequencing data becomes complex, and it require sophisticated bioinformatics tools to distinguish among overlapping genomic signatures. One more problem is faced by rare pathogens. Rare pathogens further complicate diagnostics due to limited reference databases and the potential for novel or uncharacterized genetic variations. The identification of multidrug-resistant organisms (MDOs) using NGS has been a major recent achievement, providing precise insights into resistance mechanisms and guiding targeted therapies. For example, metagenomic approaches have been successful in identifying drug-resistant Klebsiella pneumoniae and Acinetobacter baumannii strains directly from clinical samples, bypassing the need for culture-based methods [49].
When comparing NGS to conventional human identification techniques like PCR-based capillary electrophoresis (PCR-CE), significant differences arise in terms of cost, time, and efficacy. PCR-CE remains the gold standard for targeted analyses due to its affordability and rapid turnaround time, particularly in resource-constrained forensic contexts. However, it is limited by its inability to analyse highly degraded samples or detect novel allelic variations. NGS, on the other hand, offers unparalleled depth and resolution, enabling simultaneous analysis of multiple loci, which is critical for cases involving mixed DNA profiles or degraded samples often encountered in forensic investigations [50].
Despite its advantages, the adoption of NGS in routine forensic casework is hindered by high upfront costs and the need for specialized expertise. Nevertheless, advances in sequencing platforms and streamlined workflows are closing these gaps. The incorporation of hybrid approaches, where PCR-CE is used for initial screening followed by NGS for detailed analysis, is a perfect example of a cost-effective strategy. Studies have shown that while PCR-CE can quickly confirm human identity markers, NGS can provide additional data on ancestry, phenotype prediction, and even microbial signatures associated with the sample.
NGS in Forensic and Microbiology: A Game Changer
Now, let’s talk about some applications of NGS. The application of next-generation sequencing (NGS) in forensic genetics and clinical microbiology has transformed these fields by making precise and depth of analysis. In forensic genetics, NGS provides enhanced capabilities for analysing complex DNA samples, including highly degraded or mixed profiles, which are common in criminal investigations and disaster victim identification. Unlike conventional methods such as PCR-based capillary electrophoresis (PCR-CE), NGS can simultaneously examine multiple genetic loci and even mitochondrial DNA, offering a comprehensive view of genetic material in a single run. This capability allows for the detection of rare or novel alleles, ancestry markers, and phenotype predictions, which can be critical in forensic cases [49].
In clinical microbiology, NGS has proven invaluable for pathogen detection and characterization, particularly for multidrug-resistant organisms (MDOs) and rare infectious agents. By enabling whole-genome sequencing (WGS) and metagenomic analyses, NGS facilitates the identification of pathogens directly from clinical samples, bypassing traditional culture methods. This is particularly beneficial in diagnosing mixed infections or identifying novel strains, providing actionable insights for antimicrobial resistance (AMR) profiling and treatment strategies [47]. NGS thus bridges a gap in both fields, supporting researchers and practitioners with data-rich, cost-effective solutions that drive innovation in diagnostics, epidemiology, and forensic investigations.
Conclusion
High throughput sequencing has boosted research in several fields of biology and applied sciences. The utility of NGS in forensic genetics has been contested in recent years. However, applications for human identification and phenotyping are increasingly emerging. NGS offers several benefits over standard PCR-CE procedures, making it a likely future implementation in forensic laboratories. Although commercial enterprises and forensic laboratories have started the process, the present software solutions have not evolved enough. However, there are many tools or frameworks that have potentially equal or better effectiveness. These are Global Microbial Identifier which is a platform for genomic epidemiology, allowing worldwide collaboration on the storage, analysis, and comparison of whole-genome sequences of microorganisms. It supports pathogen tracking and outbreak investigations by integrating metadata and genomic data. Some other tools like Shovill and SPAdes are effective for genome assembly. Shovill provides faster assembly by optimizing Spades for larger datasets, commonly used for microbial genomics. Spades itself is a robust assembler for bacterial and viral genomes and has been enhanced for metagenomic applications [49].
High throughput sequencing has made significant progress in the past decade and is expected to continue. Improved sensitivity may lead to the replacement of PCR with probe capture technologies. Single-molecule sequencers may eventually replace NGS platforms if new signal detection benchmarks are created and base call error rates are decreased to acceptable levels.
- Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, Schloss JA, et al. (2017) DNA sequencing at 40: past, present and future. Nature. 550: 345-53.
- Kluesner MG, Nedveck DA, Lahr WS, Garbe JR, Abrahante JE, Webber BR, et al. (2018) EditR: a method to quantify base editing from Sanger sequencing. CRISPR J. 1: 239-50.
- Vincent AT, Derome N, Boyle B, Culley AI, Charette SJ (2017) Next-generation sequencing (NGS) in the microbiological world: How to make the most of your money. J Microbiol Methods. 138: 60-71.
- McCombie WR, McPherson JD, Mardis ER (2019) Next-generation sequencing technologies. Cold Spring Harb Perspect Med. 9.
- Boers SA, Jansen R, Hays JP (2019) Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur J Clin Microbiol Infect Dis. 38: 1059-70.
- Zavodna M, Bagshaw A, Brauning R, Gemmell NJ (2014) The accuracy, feasibility and challenges of sequencing short tandem repeats using next-generation sequencing platforms. PLoS One. 9: e113862.
- Weissensteiner H, Forer L, Fuchsberger C, Schöpf B, Kloss-Brandstätter A, Specht G, et al. (2016) mtDNA-Server: next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 44: W64-9.
- Legati A, Zanetti N, Nasca A, Peron C, Lamperti C, Lamantea E, et al. (2021) Current and new next-generation sequencing approaches to study mitochondrial DNA. J Mol Diagn. 23: 732-41.
- Delaney C, Garg SK, Yung R (2015) Analysis of DNA methylation by pyrosequencing. In: Immunosenescence: Methods and Protocols. 249-64.
- Kreutz M, Schock G, Kaiser J, Hochstein N, Peist R (2015) PyroMark® instruments, chemistry, and software for Pyrosequencing® analysis. In: Pyrosequencing: Methods and Protocols. 17-27.
- Miller NA, Farrow EG, Gibson M, Willig LK, Twist G, Yoo B, et al. (2015) A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med. 7: 1-16.
- Kulski JK (2016) Next-generation sequencing-an overview of the history, tools, and "Omic" applications. Next generation sequencing-advances, applications and challenges. 10: 61964.
- Heather JM, Chain B (2016) The sequence of sequencers: The history of sequencing DNA. Genomics. 107: 1-8.
- Santoferrara LF, Grattepanche JD, Katz LA, McManus GB (2014) Pyrosequencing for assessing diversity of eukaryotic microbes: analysis of data on marine planktonic ciliates and comparison with traditional methods. Environ Microbiol. 16: 2752-63.
- Slatko BE, Gardner AF, Ausubel FM (2018) Overview of next-generation sequencing technologies. Curr Protoc Mol Biol. 122: e59.
- Balloux F, Brynildsrud OB, Van Dorp L, Shaw LP, Chen H, Harris KA, et al. (2018) From theory to practice: translating whole-genome sequencing (WGS) into the clinic. Trends Microbiol. 26: 1035-48.
- Kim S, De Jonghe J, Kulesa AB, Feldman D, Vatanen T, Bhattacharyya RP, et al. (2017) High-throughput automated microfluidic sample preparation for accurate microbial genomics. Nat Commun. 8: 13919.
- Gu W, Deng X, Lee M, Sucu YD, Arevalo S, Stryke D, et al. (2021) Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med. 27: 115-24.
- Salipante SJ, Kawashima T, Rosenthal C, Hoogestraat DR, Cummings LA, Sengupta DJ, et al. (2014) Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S rRNA-Based Bacterial Community Profiling. Appl Environ Microbiol. 80: 7583-91.
- Wagner K, Springer B, Pires VP, Keller PM (2018) Molecular detection of fungal pathogens in clinical specimens by 18S rDNA high-throughput screening in comparison to ITS PCR and culture. Sci Rep. 8: 6964.
- Flurin L, Wolf MJ, Greenwood-Quaintance KE, Sanchez-Sotelo J, Patel R (2021) Targeted next generation sequencing for elbow periprosthetic joint infection diagnosis. Diagn Microbiol Infect Dis. 101: 115448.
- Hilt EE, Ferrieri P (2022) Next Generation and Other Sequencing Technologies in Diagnostic Microbiology and Infectious Diseases. Genes. 13: 1566.
- Ivy MI, Thoendel MJ, Jeraldo PR (2018) Greenwood-Quaintance KE, Hanssen AD, Abdel MP, et al. Direct Detection and Identification of Prosthetic Joint Infection Pathogens in Synovial Fluid by Metagenomic Shotgun Sequencing. J Clin Microbiol. 56.
- Niles D, Saumya D, Palazzi DL, Singh I, Revell PA (2020) Plasma Metagenomic Next-Generation Sequencing Assay for Identifying Pathogens: a Retrospective Review of Test Utilization in a Large Children's Hospital. J Clin Microbiol. 58.
- Benamu E, Gajurel K, Anderson JN, Lieb T, Gomez CA, Seng H, et al. (2021) Plasma Microbial Cell-free DNA Next-generation Sequencing in the Diagnosis and Management of Febrile Neutropenia. Clin Infect Dis. 74: 1659-68.
- Dash HR, Elkins KM, Al-Snan NR (2023) editors. Next Generation Sequencing (NGS) Technology in DNA Analysis. Elsevier.
- Khan GF, Ahad S (2018) Role of Forensic Science in Criminal Investigation: Admissibility in Indian Legal System and Future Perspective. Int J Adv Res Sci Eng. 7: 220-34.
- Sapan TU (2024) Commercial kits commonly used for NGS based forensic DNA analysis. In: Elkins KM, Zeller CB, editors. Next Generation Sequencing (NGS) Technology in DNA Analysis. Academic Press; 73-83.
- Xavier C, Parson W (2017) Evaluation of the Illumina ForenSeq™ DNA Signature Prep Kit–MPS forensic application for the MiSeqFGx™ benchtop sequencer. Forensic Sci Int Genet. 28: 188-94.
- Elkins KM, Zeller CB (2021) Next generation sequencing in forensic science: a primer. CRC Press.
- Yang Y, Xie B, Yan J (2014) Application of next-generation sequencing technology in forensic science. Genomics Proteomics Bioinformatics. 12: 190-7.
- Khodakova AS, Smith RJ, Burgoyne L, Abarno D, Linacre A (2014) Random whole metagenomic sequencing for forensic discrimination of soils. PloS One. 9: e104996.
- Bruijns B, Tiggelaar R, Gardeniers H (2018) Massively parallel sequencing techniques for forensics: A review. Electrophoresis. 39: 2642-54.
- Qian X, Hou J, Wang Z, Ye Y, Lang M, Gao T, et al. (2017) Next generation sequencing plus (NGS+) with Y-chromosomal markers for forensic pedigree searches. Sci Rep. 7: 11324.
- Wu J, Li JL, Wang ML, Li JP, Zhao ZC, Wang Q, et al. (2019) Evaluation of the MiSeqFGx system for use in forensic casework. Int J Legal Med. 133: 689-97.
- Hall CL, Zascavage RR, Sedlazeck FJ, Planz JV (2020) Potential applications of nanopore sequencing for forensic analysis. Forensic Sci Rev. 32: 23-54.
- Cornelis S, Willems S, Van Neste C, Tytgat O, Weymaere J, Vander Plaetsen AS, et al. (2018) Forensic STR profiling using Oxford Nanopore Technologies' MinION sequencer. BioRxiv. 433151.
- Cai Y, Landolfo K, Renew JR (2017) Mycobacterium infection from a cardiopulmonary bypass heater-cooler unit in a patient with steroid-induced immunosuppression. Can J Anesth. 64: 513.
- Gelardi C, Rockenbauer E, Dalsgaard S, Børsting C, Morling N (2014) Second generation sequencing of three STRs D3S1358, D12S391 and D21S11 in Danes and a new nomenclature for sequenced STR alleles. Forensic Sci Int Genet. 12: 38-41.
- Børsting C, Fordyce SL, Olofsson J, Mogensen HS, Morling N (2014) Evaluation of the Ion Torrent™ HID SNP 169-plex: a SNP typing assay developed for human identification by second generation sequencing. Forensic Sci Int Genet. 12: 144-54.
- Roewer L (2019) Y-chromosome short tandem repeats in forensics—Sexing, profiling, and matching male DNA. Wiley Interdiscip Rev Forensic Sci. 1336.
- Larsen MK, Christiansen SL, Hertz CL, Frank-Hansen R, Jensen HK, Banner J, et al. (2020) Targeted molecular genetic testing in young sudden cardiac death victims from Western Denmark. Int J Legal Med. 134: 111-21.
- Neckovic A, van Oorschot R, Szkuta B, Durdle A (2020) Challenges in human skin microbial profiling for forensic science: a review. Genes. 11: 1015.
- García MG, Pérez-Cárceles MD, Osuna E, Legaz I (2020) Impact of the human microbiome in forensic sciences: a systematic review. Appl Environ Microbiol. 86: e01451-20.
- Fungtammasan A, Ananda G, Hile SE, Su MSW, Sun C, Harris R, et al. (2015) Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res. 25: 736-49.
- Guo F, Zhou Y, Song H, Zhao J, Shen H, Zhao B, et al. (2016) Next generation sequencing of SNPs using the HID-Ion AmpliSeq™ Identity Panel on the Ion Torrent PGM™ platform. Forensic Sci Int Genet. 25: 73-84.
- Ragazzo M, Carboni S, Caputo V, Buttini C, Manzo L, Errichiello V, et al. (2020) Interpreting mixture profiles: comparison between precision ID GlobalFiler™ NGS STR panel v2 and traditional methods. Genes. 11: 591.
- Vilsen SB, Tvedebrink T, Mogensen HS, Morling N (2017) Statistical modelling of Ion PGM HID STR 10-plex MPS data. Forensic Sci Int Genet. 28: 82-9.
- Pereira R, Oliveira J, Sousa M (2020) Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. Journal of Clinical Medicine, 9(1), 132.
- Fan H, He Y, Li S, Xie Q, Wang F, Du Z, Zhu B (2022) Systematic evaluation of a novel 6-dye direct and multiplex PCR-CE-based InDel typing system for forensic purposes. Frontiers in genetics, 12: 744645.
FIGURE 1
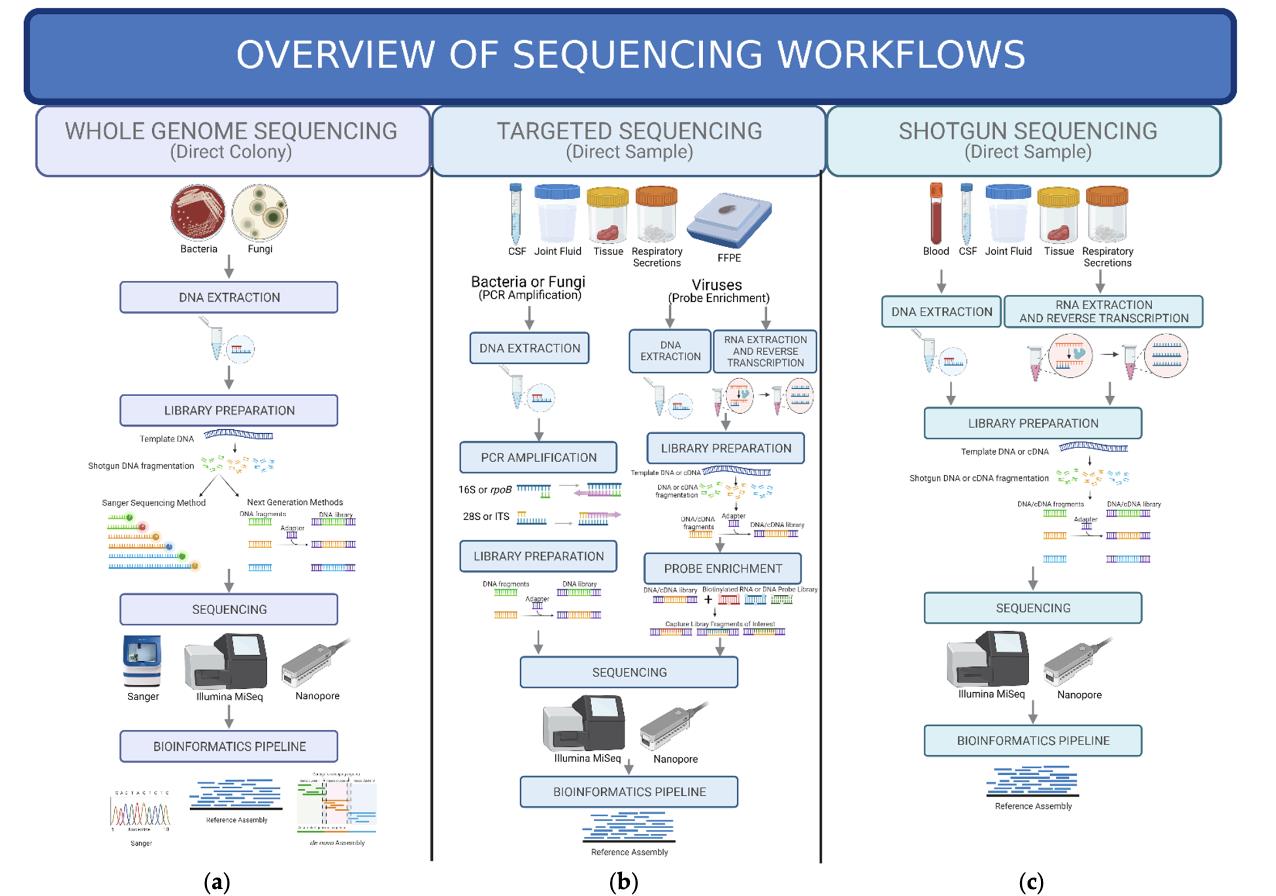
Figure 1: a) Whole Genome Sequencing: The starting is from creating a microorganism colony. DNA is extracted, fragmented, and stored in a library for Sanger sequencing or other NGS procedures. The library is sequenced and examined using bioinformatics pipelines. (b) Targeted Sequencing: This procedure starts with a clinical sample and involves selection or enrichment before library preparation for bacteria and fungus. If the pathogen of interest is a virus, selection or enrichment takes place following library preparation. After library preparation, materials are sequenced and analyzed using a bioinformatics process [22].
Figures at a glance