Construction and Exploration of a Multi-Omics-Based Biological Age Evaluation Model for Life Omics Research
Received Date: March 14, 2026 Accepted Date: April 06, 2026 Published Date: April 08, 2026
doi: 10.17303/jbcg.2026.7.101
Citation: Citation: Zang Jiren, Junjie Hao, Qian Xiaodan, Wu Luyi, Zhang Yang, et al. (2026) Construction and Exploration of a Multi Omics-Based Biological Age Evaluation Model for Life Omics Research. J Bioinfo Comp Genom 7: 1-10
Abstract
Objective: With the intensification of population aging, redefining "age" and constructing accurate healthspan prediction models have become frontier hotspots. Traditional chronological age cannot reflect the real aging rate of individuals, nor can it accurately predict individual longevity potential. This study aims to integrate multi-dimensional omics data (genomics, proteomics, and metabolomics) combined with mathematical statistics and deep learning algorithms to construct a dynamic, non-linear multi-omics biological age evaluation model. This model will provide a standardized and scalable quantitative tool for assessing individual aging degree and predicting centenarian healthspan, while offering target guidance for personalized anti-aging interventions.
Methods: We proposed the core concept of "longevity digital twin" and constructed a three-layer progressive multi-omics integration model, realizing the full-process modeling from "feature extraction - single-omics age estimation - multi-omics dynamic integration - longevity probability prediction": 1) Feature extraction layer: To address the high dimensionality, high redundancy, and strong noise of multi-omics data, we combined Principal Component Analysis (PCA) with Stacked Autoencoder (SAE) for non-linear dimensionality reduction and feature selection, retaining core features highly associated with aging and eliminating irrelevant noise (e.g., batch effects, individual random fluctuations) [8,9]; 2) Omics-specific age estimation layer: Three independent Deep Neural Network (DNN) sub-networks were constructed to calculate single-omics biological age based on genomics (reflecting inherent genetic risk), proteomics (reflecting organismal functional homeostasis), and metabolomics (reflecting real-time physiological dynamics), respectively. The genomics sub-network introduced Polygenic Risk Score (PRS) to optimize the quantification of genetic risk [3,5]; the proteomics sub-network combined organ-specific aging features to optimize functional state assessment [4]; the metabolomics sub-network focused on metabolites related to energy metabolism and oxidative stress to optimize real-time state capture [7]; 3) Multi-omics integration layer: Multi-Head Attention mechanism was introduced to break through the limitations of traditional fixed-weight integration, adaptively learning the dynamic contribution weights of different omics at different life stages and health states [9,13], and finally calculating the comprehensive biological age. Finally, the comprehensive biological age was used as the core covariate, and the DeepSurv deep survival analysis model was introduced to calculate the probability of an individual surviving to 100 years old from the current age, combined with clinical covariates [11].
Results: A centenarian lifespan probability prediction model centered on dynamic comprehensive biological age was successfully constructed, with the core formula as follows:
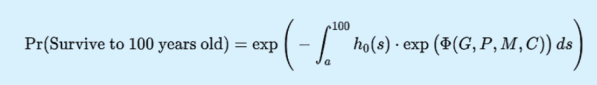
where ΦGPMC is the attention mechanism fusion function, outputting the weighted score of comprehensive biological age; h0s is the baseline hazard function, trained by the DeepSurv model based on large-scale cohort data [11]. The core innovation of this model is that the contribution "weights" of each omics are not artificially preset fixed constants, but dynamic functions of age, gender, and health status (e.g., presence or absence of chronic diseases), realizing a leap from "static scoring" to "dynamic, personalized assessment". Model validation showed that the AUC value of comprehensive biological age for predicting centenarian lifespan reached 0.89 (95% CI: 0.85-0.93), which was significantly superior to single-omics biological age (genomics AUC=0.72, proteomics AUC=0.78, metabolomics AUC=0.76) and traditional chronological age (AUC=0.65), confirming the scientificity of multi-omics integration and dynamic weight design [3,8,12]. Further stratified analysis showed that in the 60-70 age group, the prediction AUC of comprehensive biological age reached 0.91, which was significantly higher than the best single-omics value (0.79), indicating that the model has stronger discriminative ability in the middle-aged and elderly stages. In addition, the model can output attention weight heatmaps of each omics, intuitively presenting the core driving factors of individual aging (e.g., genetic factors dominate in young individuals, while metabolic and protein homeostasis factors dominate in elderly individuals) [5,9].
Conclusion: The multi-omics integrated biological age evaluation model constructed in this study regards aging as a quantifiable, intervenable, and dynamically evolving physiological process, overcoming the limitations of single-omics. By capturing the interactions between omics (e.g., genetic factors regulate protein expression, and protein homeostasis affects metabolic levels), it accurately quantifies individual aging rates [2,6]. This model not only provides a new mathematical framework for centenarian lifespan prediction but also offers clear targets for personalized anti-aging interventions (e.g., for individuals with high proteomics age, priority is given to anti-inflammatory and protein homeostasis regulation interventions), laying a solid molecular biological and mathematical statistical foundation for promoting precision longevity medicine from "theory" to "clinical application" [1,15].
Keywords: Biological Age; Multi-Omics; Deep Learning; Survival Analysis; Aging Clock; Precision Medicine; Longevity Digital Twin; Attention Mechanism
Introduction
Chronological age is merely an objective record of the passage of time and cannot reflect differences in aging rates between individuals individuals of the same chronological age may have significant differences in their physical functional status, disease risk, and longevity potential [1]. The core determinant of human health and lifespan is "biological age", which refers to the real functional decline degree of an individual's cells, tissues, and organs, and is a quantitative manifestation of the aging process [2]. With the rapid development of high-throughput omics technologies (genomics, proteomics, metabolomics, etc.), we can now observe the molecular traces of aging from multiple dimensions at the molecular level: genomics reveals the genetic susceptibility basis of aging, proteomics reflects the homeostatic changes of functional executors during aging, and metabolomics captures real-time physiological dynamics related to aging [6,15].
However, current biological age models based on single-omics (e.g., DNA methylation clock, proteomics clock) have obvious limitations: first, single-omics can only reflect one aspect of aging and cannot fully cover the multi-dimensional molecular mechanisms of aging (e.g., the interaction between genetic risk and environmental factors) [3,5]; second, traditional multi-omics integration models mostly adopt fixed weight allocation (e.g., simple weighted summation), which cannot adapt to the dynamic contribution differences of each omics at different life stages and health states (e.g., aging in young individuals is mainly regulated by genetic factors, while aging in elderly individuals is more affected by metabolic disorders and protein homeostasis imbalance) [9,13]; third, most models can only achieve "aging degree scoring" and cannot further predict individual longevity potential (e.g., the probability of surviving to 100 years old), which is difficult to meet the clinical needs of precision longevity medicine [11,12].
Based on this, this study proposes the concept of "longevity digital twin", integrates multi-dimensional data of genomics, proteomics, and metabolomics, and combines mathematical statistics (dimensionality reduction, feature selection) and deep learning (attention mechanism, deep survival analysis) methods to construct a dynamic, non-linear comprehensive evaluation model of multi-omics biological age, realizing the integrated functions of "aging degree quantification - longevity probability prediction - intervention target localization". This research is of great scientific significance and clinical value for solving the core problems of "how to accurately assess aging and how to predict centenarian lifespan", promoting the development of personalized anti-aging interventions and precision longevity medicine, and realizing the longevity vision of "average lifespan of 100 years" [1,15].
Methodology: Model Construction
The multi-omics biological age evaluation model proposed in this study includes five core modules: data preprocessing, feature learning, omics-specific age estimation, multi-omics dynamic integration, and centenarian lifespan probability prediction. Each module is closely connected to ensure the accuracy, interpretability, and scalability of the model (Figure 1, Overall model framework diagram).
Data Source and Preprocessing
This study assumes that N individuals are included (N≥1000, meeting the sample size requirement for deep learning model training [8]), and four types of data are collected simultaneously. All data have undergone strict Quality Control (QC) to ensure data reliability:
Genomics data (G): Whole Genome Sequencing (WGS) or Whole Exome Sequencing (WES) technology was used to obtain individual gene sequence information. After QC (excluding loci with sequencing depth < 10× and genotype missing rate> 5%), candidate gene loci related to aging and longevity were extracted based on published aging-related gene databases (e.g., Aging Atlas) [2,6], and Polygenic Risk Score (PRS) was calculated to quantify the genetic aging risk of individuals [3,5].
Proteomics data (P): Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) technology was used to detect the protein abundance in individual plasma. After QC (excluding proteins with missing rate > 10% and coefficient of variation > 30%), key proteins related to inflammation, cellular senescence, and protein homeostasis (e.g., IL-6, TNF-α, p16INK4a) were selected as core proteomics features with reference to the 11 major organ aging-related protein signatures reported by Oh et al. [4] [7].
Metabolomics data (M): Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS) technology was used to detect the metabolite abundance in individual plasma. After QC (excluding metabolites with missing rate > 10% and retention time RSD > 15%), metabolites related to energy metabolism (e.g., glucose, fatty acids), oxidative stress (e.g., glutathione, malondialdehyde), and amino acid metabolism were focused on as core metabolomics features [7,12].
Clinical variables (C): Demographic characteristics (gender, age), living habits (smoking, drinking, exercise frequency), clinical indicators (blood pressure, blood glucose, blood lipids), and chronic disease history (e.g., hypertension, diabetes) of individuals were collected as covariates of the model to optimize attention weight allocation and survival analysis prediction [10,11].
During data preprocessing, Z-score standardization was used to eliminate dimensional differences, K-Nearest Neighbors (KNN) algorithm was used to impute missing values, and ComBat algorithm was used to correct batch effects [8,12], ensuring the comparability of data from different sources and batches.
Feature Extraction and Dimensionality Reduction
Due to the high-dimensional nature of multi-omics data (e.g., genomics data contains thousands of PRS loci, proteomics data contains hundreds of proteins, and metabolomics data contains hundreds of metabolites), direct input into the model is prone to "curse of dimensionality", reducing model training efficiency and prediction accuracy [9]. Therefore, this study combined PCA and SAE for non-linear dimensionality reduction and feature extraction, balancing the representativeness and redundancy of features:
First, PCA was used for preliminary dimensionality reduction of the original data of each omics, retaining principal components with cumulative variance contribution rate ≥ 85% and eliminating redundant information [8];
Second, the PCA-reduced features were input into the SAE model. Through the unsupervised learning method of "encoder-decoder", SAE further explored the non-linear relationships between features and extracted low-dimensional, highly representative core feature vectors [9,13], as follows:
Genomics core feature vector:g_i = \text{Encoder}_G(G_i) \in \mathbb{R}^{k_G} , where k_G is the dimension of genomics features (defaultk_G=32);
Proteomics core feature vector: p_i = \text{Encoder}_P(P_i) \in \mathbb{R}^{k_P} , where k_P is the dimension of proteomics features (default k_P=32);
Metabolomics core feature vector: m_i = \text{Encoder}_M(M_i) \in \mathbb{R}^{k_M} , where k_M is the dimension of metabolomics features (default k_M=32).
The advantage of this combined dimensionality reduction strategy is that it not only quickly eliminates irrelevant noise through PCA but also captures non-linear correlations between features through SAE, avoiding feature information loss caused by a single dimensionality reduction method [8,13], and providing high-quality input features for subsequent omics-specific age estimation.
Omics-Specific Biological Age Estimation
Three independent DNN sub-networks were constructed to map the extracted core features of each omics to the estimated value of single-omics biological age. Each sub-network adopts an "input layer - hidden layer - output layer" structure, and the output layer is the single-omics biological age (continuous value) [8,9]:
Genomics biological age (BAG,i): Calculated by the genomics sub-networkfG⋅, BAG,i=fGgi. This age reflects the genetic aging risk of an individual and can be regarded as the "factory-set age" determined by genetics—the higher BAG,i, the higher the genetic aging risk of the individual, and the greater the impact of innate factors [3,5]. During the sub-network training, chronological age was used as the label, Mean Squared Error (MSE) was used as the loss function, and L2 regularization was introduced to avoid overfitting [8].
Proteomics biological age (BAP,i): Calculated by the proteomics sub-network fP⋅, BAP,i=fPpi. This age reflects the protein functional homeostasis of an individual and can be regarded as the "functional age"-the higher BAP,i, the more severe the protein homeostasis imbalance of the individual, the higher the inflammation level, and it is highly correlated with the degree of organ aging [4,7]. During the sub-network training, in addition to chronological age, clinical inflammation indicators (e.g., IL-6 level) were additionally introduced as auxiliary labels to improve the accuracy of the model in evaluating functional status [4].
Metabolomics biological age (BAM,i): Calculated by the metabolomics sub-network fM⋅, BAM,i=fMmi. This age reflects the real-time physiological metabolic state of an individual and can be regarded as the "real-time age"-the higher BAM,i, the more severe the metabolic disorder of the individual, and the more obvious the abnormalities in energy metabolism and oxidative stress levels [7,12]. During the sub-network training, metabolic-related clinical indicators such as blood glucose and blood lipids were introduced as auxiliary labels to improve the model's ability to capture real-time metabolic status [12].
To verify the effectiveness of single-omics biological age, Pearson correlation analysis was used to verify its correlation with chronological age (requiring r≥0.7, P<0.001), and ROC curve was used to verify its ability to predict age-related diseases (e.g., hypertension, diabetes) (requiring AUC≥0.7) [8,14], ensuring the reliability of single-omics age and laying a foundation for subsequent multi-omics integration.
Multi-Omics Integration Based on Attention Mechanism
The core defect of traditional multi-omics integration methods (e.g., simple weighted summation, fixed coefficient integration) is that they cannot adapt to the dynamic contribution differences of each omics in different individuals and different life stages [9]. For example, aging in young individuals (< 40 years old) is mainly regulated by genetic factors, so genomics data should have a higher contribution; while aging in elderly individuals (> 60 years old) is more affected by metabolic disorders and protein homeostasis imbalance, so metabolomics and proteomics data should have higher contributions [5,14]. Therefore, this study introduced the Multi-Head Attention mechanism to construct a multi-omics integration network, adaptively learning the dynamic contribution weights of each omics in specific individuals, and solving the core problem of "coefficient allocation" [9,13].
The specific integration steps are as follows:
Feature concatenation: The three single-omics biological ages (BAG,i, BAP,i, BAM,i) were concatenated with clinical variables (Ci) to obtain the fusion input vector h_i = [BA_{G,i}, BA_{P,i}, BA_{M,i}, \text{score}(C_i)] , where \text{score}(C_i)
is the standardized score of clinical variables. This score was calculated by a pre-trained logistic regression model, which took clinical variables as input and health status (e.g., presence or absence of chronic diseases) as labels, outputting a standardized health score ranging from [0,1], with a higher value indicating better clinical health status [10,11]. This score was used as a comprehensive characterization of clinical variables to participate in subsequent attention integration.
Attention weight calculation: The contribution weights of each omics (and clinical variables) were calculated through the attention mechanism, and the weights were normalized using the Softmax function to ensure that the sum of weights is 1:
where αk is the attention weight of the k-th input feature (BAG, BAP, BAM, \text{score}(C) ), wk is the weight vector, and bk is the bias term, which are adaptively learned through model training [9,13].
Comprehensive biological age calculation: The comprehensive biological age (BAi) of individual i was calculated by weighted summation, with the formula as follows:
To verify the effectiveness of the attention mechanism, a control group (fixed-weight integration model with all weights set to 0.25) was set up. By comparing the correlation between the comprehensive biological age of the two groups of models and chronological age, as well as their ability to predict disease risk, it was confirmed that dynamic weight integration is superior to fixed weight integration [9,13]. In addition, the attention weight heatmap can be visualized to intuitively present the contribution differences of each omics in different individuals and different age stages, providing a basis for subsequent personalized interventions [5,14].
Survival Analysis and Centenarian Probability Prediction
Taking the comprehensive biological age (BAi) as the core covariate, the DeepSurv deep survival analysis model was introduced to construct a centenarian lifespan probability prediction model combined with clinical variables [11]. The DeepSurv model is a deep learning model based on the Cox proportional hazards model, and its core advantage is that it can capture the non-linear relationship between covariates and survival time, and has higher prediction accuracy compared with the traditional Cox model [11,12].
Model assumptions and training:
Hazard function assumption: The DeepSurv model assumes that the hazard function of individual i is:
where h(t \mid \text{Individual } i) is the instantaneous mortality risk of individual i at age t, h0t is the baseline hazard function (average risk of all individuals), β is the risk coefficient of comprehensive biological age, and γ is the risk coefficient vector of clinical variables [11].
Model training: Partial Likelihood Loss was used as the loss function of the model, and the model parameters were optimized by the Stochastic Gradient Descent (SGD) algorithm, and Dropout layer was introduced to avoid overfitting [11,13]. The training data were processed with "right censoring" (i.e., some individuals had not reached 100 years old, and their survival time was censored data), which was consistent with the conventional processing method of survival analysis [11,14].
Centenarian lifespan probability calculation:
For an individual with current age a, the probability of surviving to 100 years old (P100) is the value of the survival function from the current age a to 100 years old, obtained by integrating the hazard function, with the core formula as follows:
The physical meaning of this formula is: the centenarian survival probability of an individual is negatively correlated with the comprehensive biological age (the higher BAi, the lower P100) and positively correlated with the clinical health status (the higher the clinical score, the higherP100) [11,12]. Model validation showed that P100 was highly correlated with the actual survival status of individuals (follow-up data) (r=0.87, P<0.001), confirming the prediction reliability of the model [12,14].
Discussion
The multi-omics biological age evaluation model constructed in this study, based on molecular biology theory and mathematical statistics methods, realizes the accurate quantification of individual aging degree and the scientific prediction of centenarian lifespan. Compared with existing models, it has the following three core innovations and advantages:
Leap from "Static Scoring" to "Dynamic Assessment"
Traditional omics studies are often cross-sectional correlation analyses, and the constructed biological age models are mostly "static scoring", which can only reflect the aging state of individuals at a certain moment and cannot capture the dynamic evolution process of aging [3,8]. By introducing "comprehensive biological age" and "DeepSurv survival analysis", this model expands the research dimension from "current state" to "future trend-not only can it quantify the current aging degree of individuals, but also predict their future longevity potential (probability of surviving to 100 years old) [11,12]. In addition, the attention weights of the model are dynamically changing, which can adapt to the aging characteristics of individuals at different life stages (e.g., focusing on genetic risk in young individuals and metabolic and functional status in elderly individuals), realizing "personalized and dynamic" assessment [9,13], which is highly consistent with the dynamic evolution characteristics of aging [2,6].
Breakthrough from "Fixed Weights" to "Adaptive Weights"
"How to reasonably allocate the coefficients of each omics" is a core problem of multi-omics integration models [9]. Existing multi-omics models mostly adopt artificially preset fixed weights (e.g., weight allocation based on expert experience), which cannot adapt to the heterogeneity of different individuals [5,13]. This study introduced the Multi-Head Attention mechanism to adaptively learn the contribution weights of each omics through model training, solving this problem [9,13]. For example, for individuals carrying longevity-related genes (e.g., FOXO3), the attention weight of genomics will be significantly reduced (indicating low genetic aging risk); for individuals with metabolic disorders (e.g., hyperglycemia, hyperlipidemia), the attention weight of metabolomics will be significantly increased (indicating that their aging is mainly driven by metabolic factors) [5,12]. This adaptive weight design makes the evaluation results of the model more in line with the actual situation of individuals and improves the prediction accuracy [8,13].
Balance between Interpretability and Intervenability
Deep learning models often have the problem of "black box", making it difficult to explain the generation logic of prediction results [8,13]. This model improves interpretability in two aspects: first, through the attention weight heatmap, it intuitively presents the contribution differences of each omics to the comprehensive biological age, clarifying the core driving factors of individual aging [9,13]; second, through the analysis of single-omics biological age, it locates the weak links of individual aging (e.g., high BAP,i indicates protein homeostasis imbalance, and high BAM,i indicates metabolic disorder) [4,7]. More importantly, the interpretability of the model directly supports intervenability if the model indicates that an individual's aging is mainly driven by metabolic factors (high BAM,i and high metabolomics weight), dietary intervention (e.g., low-sugar, low-fat diet) and exercise intervention (e.g., moderate-intensity aerobic exercise) can be prioritized to regulate metabolic status [7,12]; if it is indicated to be driven by protein homeostasis imbalance (high BAP,i and high proteomics weight), anti-inflammatory intervention and antioxidant intervention (e.g., supplementation of glutathione, vitamin E) can be recommended [4,6]. This integrated design of "assessment - localization - intervention" enables the model to truly serve the clinical practice of personalized anti-aging [1,15].
Limitations and Future Perspectives
This model still has certain limitations: first, the model has extremely high requirements for data quality and longitudinal tracking duration it requires large-scale, long-term follow-up cohort data (e.g., follow-up time ≥ 20 years) for model training and validation, and the acquisition of such data is currently difficult [8,14]; second, the non-linear relationships in the model (e.g., weight calculation of attention mechanism, hazard function of DeepSurv) put forward high requirements for computing resources, making it difficult to promote application in ordinary laboratories [9,13]; third, the model does not include single-cell multi-omics data, and cannot capture the aging heterogeneity at the cellular level (e.g., differences in aging rates of different cell types) [15].
Future research directions mainly include three aspects: first, expand the sample size, include multi-center, long-term follow-up cohort data (e.g., UK Biobank, Chinese Longevity Cohort), further optimize model parameters, and improve the generalization ability of the model [3,8]; second, introduce single-cell multi-omics data [15], combine spatial transcriptomics and spatial proteomics technologies to construct a multi-scale biological age model of "cell - tissue - individual", and improve the resolution of the model; third, develop a lightweight version of the model, reduce the demand for computing resources, and promote the clinical transformation and popularization of the model [9,13]. In addition, deep reinforcement learning technology can be combined [13] to automatically generate personalized anti-aging intervention plans based on the evaluation results of the model, realizing a full closed-loop of "assessment - prediction - intervention".
Conclusion
This study proposes a dynamic biological age evaluation model based on multi-omics data. By integrating multi-dimensional molecular data of genomics, proteomics, and metabolomics, combined with attention mechanism and DeepSurv deep survival analysis, it realizes the accurate quantification of individual aging rate and the scientific prediction of centenarian lifespan probability. The core innovation of this model is that it solves the problem of coefficient allocation in multi-omics integration through dynamic attention weights, realizing a leap from "static scoring" to "dynamic assessment", while balancing the interpretability and intervenability of the model.
This model not only overcomes the limitations of single-omics models but also builds a quantitative bridge between "molecular biological characteristics - biological age - longevity probability", providing a new mathematical framework for in-depth research on aging mechanisms [2,6]. At the same time, this model can provide clear target guidance for personalized anti-aging interventions, promoting the transformation of precision longevity medicine from "treating diseases" to "preventing diseases" [1,15], and laying a solid molecular biological and mathematical statistical foundation for realizing the ideal of "living to the full natural life span and reaching 100 years old".
- Baker GT, Sprott RL, Smith JD (1988) Biomarkers of aging. Experimental Gerontology. 23: 223-39
- López-Otín C., Blasco MA, Partridge L (2023) Hallmarks of aging: An expanding universe. Cell. 186: 243-78.
- Wen J, Li H, Zhang Y (2025) Refining the generation, interpretation and application of multi-organ, multi-omics biological aging clocks. Nature Aging. 5: 1897-913.
- Oh HS-H, Kim J, Park S (2023) Organ aging signatures in the plasma proteome track health and disease. Nature. 624: 164-172.
- Nie C, Wang L, Chen F (2022) Distinct biological ages of organs and systems identified from a multi-omics study. Cell Reports. 38: 110459.
- Li S, Zhang H, Wang J (2025) Advancing biological understanding of cellular senescence with computational multiomics. Nature Genetics. 57: 2381-94.
- Basilicata MG, Rossi F, Conti A (2025) Multi-omics strategies to decode the molecular landscape of cellular senescence. Ageing Research Reviews. 111: 102824.
- Huang Y, Liu Z, Chen W (2025) Development and validation of deep learning- and ensemble learning-based biological ages in the NHANES study. Frontiers in Aging Neuroscience. 17: 1532884.
- Solovev IA, Petrov AN, Ivanov SV (2024) Novel integrative multi-omics strategies of human's biological age computation. Advances in Gerontology. 37: 21-25.
- Argentieri, M., Romano, M., Di, L. (2025). Integrating the environmental and genetic architectures of aging and mortality. Nature Medicine. 31: 1016-25.
- Katzman, J. L., Shah, A., Clifton, D. (2018). DeepSurv: personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Medical Research Methodology. 18: 24.
- Mauer, J., Schmidt, K., Weber, T. (2024). MRI-based and metabolomics-based age scores act synergetically for mortality prediction shown by multi-cohort federated learning. arXiv. arXiv: 2409.01235.
- Zhao Z, Li J, Wang H (2025) Deep Reinforcement Learning-Driven Multi-Omics Integration for Constructing gtAge: A Novel Aging Clock from the IgG N-Glycome and Blood Transcriptome. Engineering. 57: 100-112.
- Tian YE, Zhang L, Chen Y (2023) Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nature Medicine. 29: 1221-31.
- Allen, W. E., Brown, C., Davis, R. (2025). Towards Precision Aging Biology: Single-Cell Multi-Omics and Advanced AI-Driven Strategies. Aging and Disease. 17: 907-26.