A Novel AZIN1 Targeted Multiepitope Therapeutic Vaccine Designed for Non-Small Cell Lung Carcinoma Using Immunoinformatics Approach
Received Date: May 18, 2026 Accepted Date: June 07, 2026 Published Date: June 12, 2026
doi: 10.17303/jbcg.2026.7.102
Citation: Seema Kalra, Anita Chauhan, Rashmi Kumari (2026) A Novel AZIN1 Targeted Multiepitope Therapeutic Vaccine Designed for Non-Small Cell Lung Carcinoma Using Immunoinformatics Approach. J Bioinfo Comp Genom 7: 1-22
Abstract
Non-small cell lung cancer (NSCLC) is one of the most prevalent and fatal cancer worldwide. Vaccines being used currently for NSCLC treatment are based on single-target with low specificity and don’t not show promising results. To address this limitation, we have designed a multi-epitope vaccine using immunoinformatics approach. The study was aimed to design a multi-epitope subunit vaccine that targets the AZIN1 protein, a novel target for non-small cell lung carcinoma (NSCLC). The safety and efficacy of the vaccine was predicted by studying its physicochemical properties, molecular docking, thermodynamics stability profiling, immune simulation and codon optimization. Different tools from IEDB server and BCPRED were employed to predict B- and T-cell epitopes within antigenic AZIN1 protein followed by the selection of most immunodominant peptides having overlapping B and T cell epitopes. Since, VaxiJen revealed a significant antigenicity score for the sequence, therefore, a vaccine of immunogenic peptides specific to NSCLC was designed by molecular modeling and simulation studies using WebGRO-UAMS and C-ImmSim Server. The constructed vaccine has overlapping T-cell and B-cell epitopes from AZIN1 protein that are highly antigenic, non-allergenic, non-toxic, conserved, and non-homologous. Molecular docking of antibodies with human TL- receptors showed strong affinity for these receptors and the predicted vaccine stimulated an excellent immune response. Codon optimization revealed its expression potential (CAI value 0.959). The AZIN1 based predicted vaccine is a novel and promising candidate considering its safety, efficacy in humans as shown in this in silico study.
Keywords: Non-Small Cell Lung Carcinoma; Antizyme Inhibitor 1; In Silico Vaccine Design; Epitope Prediction; Molecular Dynamic Simulation; Immunoinformatics
Introduction
Lung cancer is the most commonly diagnosed cancer second only to breast cancer. Non-small cell carcinoma (NSCLC) being asymptomatic at early stages is diagnosed only at advanced stages, which limits the therapeutic options for the patients [1].
Resection of cancer tissues in patients at an advanced stage NSCLC is not possible; therefore, different treatment routines are followed to treat such patients. Chemotherapy, radiotherapy and metal-based therapies with some combination of drugs are part of the treatment. Although these therapies increase the survival to some extent but overall outcome is not impressive [2]. Various drugs based on biomarkers EGFR, ALK, AKT, RAS, BRAF, PD1 and CTLA4 are used for NSCLC treatment [3]. A brief analysis of these biomarkers is given in Table 1.
Despite new developments in the treatments of patients diagnosed with advanced NSCLC, many challenges persist. One important challenge is resistance to the drug of treatment after 6-7 months of use [8]. A possible reason may be use of single target-based biomarkers; as cancer involves multiple genes and proteins. Moreover, cancerous cells recruit alternative pathways to skip the single target or the target itself may get mutated [9, 10]. Therefore, poor performance of these therapies for treatment NSCLC has always kept the researchers exploring for the ways to develop new therapies for NSCLC [11].
After continuous disappointments from single target therapies for NSCLC, multi target therapies using systems biology and immunoinformatics approaches are becoming more and more important. Today, the clinical focus is on developing efficient therapeutic cancer vaccines that are able to promote an effective and durable T cell response to specific tumor antigens.
Antizyme inhibitor 1 (AZIN1) has emerged as an important gene which plays an important role in many cancers including NSCLC. It regulates polyamine biosynthesis which promotes cell differentiation, growth and proliferation. Many studies prove that increased RNA editing of AZIN1in tumors leads to mutations and its overexpression. Its dysregulated interactions promote tumor growth, differentiation and stemness in both polyamine dependent and independent pathways [12, 13]. Mutations in AZIN1 has critical role in cancer cell migration mainly by upregulation of the IL-8 [14]. In our previous study, we showed that AZIN1 is the hub gene of dynamic network biomarker (DNB) of five genes (DNTTIP1, EIF3H, HMGCR, PGAM1 and AZIN1) which could play an important role in NSCLC metastasis. Functional enrichment studies indicated the role of these DNB genes in purine metabolism. In silico analysis of different stages of NSCLC revealed large changes in expression of DNB genes at stage 2A-2B pointing it to be the tipping point of metastasis. Further, suppression of AZIN1 expression by miRNA silencing altered the expression of other genes of DNB in NCI-40 cell line. These results indicated dysregulated expression of AZIN1 DNB genes might play role in NSCLC metastasis [15].
Prominent role of AZIN1 in cancers, its reported mutations and over expression in tumors makes it an interesting candidate for tumor associated antigen that could be explored for a potential vaccine design. The study was designed having objectives: (i) to predict B -cell and T-cell epitopes with AZIN1 as target; (ii) to design a multi-epitope subunit vaccine to target the AZIN1 protein; (iii) to predict the safety and efficacy of the vaccine by studying physicochemical properties, molecular docking, thermodynamic stability profiling; and (iv) to analyze immune simulation and codon optimization. Flowchart of the study is given in Fig.1.
Materials and Methods
Protein Sequence Retrieval
Structure of human antizyme inhibitor (PDB ID: 4ZGZ) was retrieved from PDB (http://rscb.org/). The FASTA sequence of this protein was retrieved from the National Center of Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/).
B cell Epitope Prediction
From the sequence of AZIN1, B-cell epitopes were predicted by several B-cell prediction methods in IEDB. Different methods include BepiPred [16], Chou & Fasman Beta-Turn Prediction [17], Emini Surface Accessibilty Prediction [18] , Karplus & Schulz Flexibility Prediction [19], Kolaskar and Tongaonkar Antigenicity [20] and Parker Hydrophilicity Prediction [21] with default window and threshold values.
BepiPred uses hidden Markov model and a propensity scale method to predict the location of linear B-cell epitopes. The residues with scores above the threshold (threshold value is 1) are predicted as a part of the epitope and colored in yellow on the graph (where Y-axis depicts residue scores and X-axis residue positions in the sequence) and marked with "E" in the output table. Chou Fasman predicts turns and antigenicity of the epitopes were predicted by Kolarkar method. Parker method identified the hydrophilicity of the residues.
Cytotoxic T-Cell Epitope Predictions
To predict T-cell epitope, MHC (major histocompatibility complex) alleles for HLA class I and HLA class II in human population were selected. IEDB predicts MHC class I epitopes of length 9 amino acids based on the IC50 values (<50nM) of peptide. Cytotoxic T lymphocytes (CTLs) epitopes were predicted as they have crucial role in cancers. Next, these predicted epitopes were subjected to IEDB MHC-I prediction tool. 10 immunogenic epitopes having IC50 < 50 nM and VaxiJen antigenicity score> 0.4 were predicted.
Helper T-Cell Epitope Predictions
MHC II binding epitopes were analyzed using IEDB (http://tools.immuneepitope.org/mhcii/). The structure of MHC II has a deep groove which can accommodate a long sequence of 15 amino acids. Therefore, peptide length was set at 15 amino acids. 10 Epitopes were selected based on IC50 value (<50 nM)) and VaxiJen antigenicity score > 0.4.
Due to allelic variation in individuals, it is not possible to treat all with a single epitope. Therefore, it is evident to combine multiple epitopes or to take overlapping immunodominant regions containing both B cell and T cell epitopes. In the present study, multiple sequence alignments (MSAs) tool Clustal Omega was used for aligning the B cell and T cell epitopes which align big sequences quickly and accurately [22].
Population Coverage Analysis
Population coverage analysis for individuals is crucial because epitopes may show allelic variations during interaction with different HLA alleles. Therefore, IEDB was employed to find out the percentage of people expected to respond to a specific number of MHC-restricted epitopes in Asian population and worldwide.
Multi-Epitope Vaccine Construction
Sequence of adjuvant was retrieved from NCBI. An adjuvant PADRE was attached to N terminal of vaccine whereas EAAAK and AAY were used to construct vaccine sequence [23].
Prediction of Secondary Structure and Physiochemical Analysis
The secondary structure of a protein is an essential component in determining its folding as well as physicochemical properties. Thus, for the prediction of the secondary structure of our constructed multi-epitope vaccine and to assess its structural properties and elements, we used the tool ProtParam (https://web.expasy.org/protparam/) [24].
The ProtParam allows the computation of grand average hydropathicity parameters (GRAVY), estimated half-life, molecular weight, instability and aliphatic index, theoretical pI, amino acid and atomic composition, and extinction coefficient of the given protein. The GRAVY value of protein is calculated by adding up the hydropathy values of all of the amino acids, dividing the total by the number of residues in the sequence.
Prediction of Peptide Allergenicity, Transmembrane Topology and Solubility
Vaccine antigens have the potential to cause allergic reactions; even mild reactions can lead to severe complications. Unveiling allergenicity of peptides is an important consideration to avoid such conditions. AlgPred [25] checks peptide allergenicity of peptides by three methods, IgE binding, MAST and Blast allergen search. TMHMM server (http://www.cbs.dtu.dk/services/TMHMM/.) was used to check the Transmembrane (TM) spanning regions of the predicted vaccine candidate.
Tertiary Structure Prediction of the Vaccine
SWISS Model [26] predicted three-dimensional (3D) structure of the vaccine. SWISS-MODEL is a web-based integrated service dedicated to protein structure homology modelling. It guides the user to building protein homology models at different levels of complexity. GMQE score gives an overall model quality measurement between 0 and 1. Higher value indicates high expected quality. Small structural distortions, unfavourable interactions or clashes introduced during the structure modelling process are resolved by energy minimization and a clash score is calculated. Clash score is an approximation of the overall severity of the clashes in a structure. The molprobity score provides a single number that represents the protein quality statistics.
Molecular Dynamics Fluctuation Analysis
All the toll-like receptors (TLRs) and peptide vaccine complex were subjected to docking, refining and dynamic studies to monitor the stability of the vaccine construct followed by the root mean square fluctuation graphs.
Molecular docking between the TLRs and the designed vaccine was done by an online server HDOCK (http://hdock.phys.hust.edu.cn/). It is an integrated server for robust protein–protein and protein -DNA docking. The server delivers both template- and docking-based binding models of two molecules and allows for download and interactive visualization.
Molecular Dynamics Simulations
Simulation studies were done using WebGro. It is an automated online tool for analysing dynamics simulation of proteins alone or in complex with ligands. It is based on the GROMACS simulation for performing molecular dynamics simulations in solvents as well as trajectory analysis [27].
Immune Infiltration Analysis
The composition and abundance of immune cells in the tumor microenvironment defines how patient will respond to tumor progression and immunotherapy. Direct measurement methods have limitations in predicting the abundance of immune cells. Therefore, computational algorithms are often used to infer immune system information from bulk tumor transcriptome profiles [28, 29].
For analyzing immune infiltrates in different cancer types TIMER 2.0 (Tumor Immune Estimation Resource) database (http://timer.cistrome.org/) was used [30]. The results for the correlation between our hub gene AZIN1 and tumor immune check points PD-1, PD-L1 and CTLA4 were analysed considering statistically significant at P value<0.05. The Spearman’s coefficient was used to calculate the correlation between AZIN1 expression and immune cells.
Immune Simulation
To detect the immune response of the predicted vaccine, the C-ImmSim Server (https://kraken.iac.rm.cnr.it/C-ImmSim) was used to perform the immune simulation of cellular and humoral immunity of vaccine to mammals [31]. The injection procedure was at time steps equal to 1 (one time step corresponds to 8h). Other parameters like Random Seed, the Simulation Volume, the Adjuvant dose and Number of antigens injected were system default [32].
By adjusting the host organism's codon bias, codon optimization is used to enhance the efficiency of translation and encourage the expression of a gene of interest. Codon bias is employed to achieve this [33]. Avoiding restriction enzyme locations was part of the codon optimization procedure. Results for the Codon adaptation index (CAI) and GC content percentage were also acquired. The vaccine construct was ultimately subjected to reverse translation, producing a 150-nucleotide cDNA sequence. The codon was adjusted in relation to the process of gene expression in Homo sapiens using JCat [34].
Results
Prediction of B Cell Epitopes
As B-cell epitopes play an important role in vaccine design, clinical diagnosis, and antibody production, a limited number of prediction models for linear or conformational B-cell epitopes have been developed.
From the sequence of AZIN1, B-cell candidate epitopes were analysed by several B-cell prediction methods from IEDB including BepiPred with threshold>0.5, Chou & Fasman Beta-Turn Prediction, Parker Hydrophilicity Prediction Karplus & Schulz Flexibility Prediction, Emini Surface Accessibility Prediction and Kolaskar & Tongaonkar Antigenicity with default window and threshold values (Fig.2A-F).
The hydrophilicity of anticipated B-cell epitopes was studied using the Parker-hydrophilicity analysis. The Chou and Fasman scale predicts beta turns. Surface accessibility of epitope was predicted by
Emini method it is calculated as surface probability (Sn) = (n+4+i ) (0.37)-6 where Sn stands for fractional surface probability value, and its value varies from 1 to 6. B-cell linear epitopes were identified by comparing results of predictions of all these algorithms. Only those top five epitopes were selected which were predicted by more than four methods. When subjected to VexiJen test for antigenecity, these peptides passed the test and has threshold score greater than 0.4 (Fig. 3).
Prediction of Cytotoxic T Lymphocyte Epitopes
Cells have antigen presenting cells on the surface. T-cell receptors (TCR) recognize T-cell epitopes and MHC molecules present them. Antigens presented on MHC class I molecules are recognized by Cytotoxic TCRs while antigens presented on MHC class II molecules are recognized by helper T-cell. Humans consist of three MHC class I genes namely HLA-A, HLA-B, and HLA-C and three MHC class II genes; HLA-DR, HLA-DQ, and HLA-DP. The distribution of HLA genes varies in the worldwide population. Epitopes which bind to MHC class I molecules are short in size in the range 8-15 amino acids whereas MHC class II molecules can bind to longer peptides of length 11 to 30 amino acids. CTL epitopes were predicted using IEDB MHC-I prediction tool. 10 immunogenic epitopes each being 9 amino acid long were selected based on their IC50 value < 50nM and VaxiJen score > 0.4 (Fig. 4).
Out of all the predicted epitopes, the epitope ‘244FQLEEVNHV252’ interacting with HLA-A*02:06 allele had highest affinity and eptiope ‘61AQIKPFYIV69’ interacting with HLA-A*02:06 allele had lowest affinity.
Prediction of Helper T Lymphocyte Epitopes
To select MHC II binding epitopes, peptide length was set at 15 amino acids with IC50 value (<50 nM). All the epitopes maintained their IC50 less than 50 nM and VaxiJen antigenicity score greater than 0.4 (Fig 5).
Selection of Immunodominant Regions
As discussed earlier, allelic sequence of HLAs vary in a population or region. Therefore, providing antigenic protection using one epitope is difficult. To address this issue, immunodominant regions containing overlapping B cell and T cell epitopes were selected as these might show an effective immune response. Sequence alignment of twenty T-cell and five B- cell epitopes helped to identify two immunodominant peptides having overlapping B- cell and T- cell epitope regions (Fig.6). These two peptides were ‘VSQIKYFYTVKCNSAPAVL’ and ‘NAFFVGDLGKIVKKH’.
Prediction of Population Coverage
Population coverage analysis for the selected epitopes demonstrated the consequences of the host genetic variation on the binding specificity of these targeted epitopes to class I HLA alleles. Their cumulative population coverage around the Asian continent was 90%, which revealed that these epitopes could cover about 90% of the population from different regions of the Asian continent (Fig. 7).
Multi-Epitope Vaccine Construction and Physicochemical Analysis
The secondary structure of a protein determines its folding and physicochemical properties. Thus, to predict the secondary structure of the multi-epitope vaccine construct and assess its structural properties, ProtParam was used. The criteria for potential epitopes for construction of the linear vaccine included: (a) antigenic and non-allergic; (b) high binding affinity to MHC alleles; (c) promiscuous; (d) presence of overlapping CTL and HTL epitopes. Two epitopes ‘VSQIKYFYTVKCNSAPAVL’ and ‘NAFFVGDLGKIVKKH’ fulfilled these criteria and were found suitable to construct a linear vaccine sequence.
For the construct, all CTL, HTL, and B-cell sequences were grouped, regardless of the order in which the amino acid sequences appear. However, short chains of amino acids known as linkers are required to separate the different sequences. Although they serve different functions, linkers KK, AAY, GPGPG, and EAAAK are all often utilized. HTL, CTL, and B cell epitopes are typically joined by AAY, KK, and GPGPG linkers, respectively. There are several benefits to using AAY and EAAAK linkers, including the prevention of junctional epitope development and the facilitation of epitope separation, which makes the epitopes accessible for immune processing [35].
To increase protein stability, the EAAAK linker keeps domains apart. The EAAAK sequence when used should come right after the adjuvant sequence. Since many effective adjuvants cannot be represented as amino acid sequences, there are currently few alternatives for in silico adjuvants. In silico vaccine efficacy evaluation techniques rely on FASTA sequences, which restrict the set of requirements that must be met. An adjuvant is used to boost immunological responses. An adjuvant was added to the vaccine design at the N-terminal to increase immunogenicity (Fig. 8).
PADRE, a synthetic 13 amino acid peptide that aids in CD4+ T cell activation, was employed as an adjuvant. PADRE (AKFVAAWTLKAAA) was chosen because of its strong binding to 15 of the 16 most prevalent human HLA-DR types. Consequently, in any population, PADRE can overcome the polymorphism issue related to HLA-DR molecules. A study by Wu et al. (2010) showed that PADRE is safe and well tolerated in humans and is 100-fold more potent than other universal T helper epitopes [36].
The total number of amino acids in the construct was 55. GRAVY score was 0.407 and protein was stable having instability index 10.12. The instability index threshold for proteins is less than 40.
Modeling of Tertiary Structure
The Swiss Model was used to forecast the 3D model of the vaccine construct (Fig. 9). The model had a Global Model Quality Estimate (GMQE) of 0.7. The model quality score ranges from 0 to 1. The anticipated model's Molprobity of 0.96 was suitable for normal structures. The number of conflicts per 1,000 atoms, including hydrogen indicated by the clash score was 1.93.
100% favorable residues were predicted using the Ramachandran plot (Fig.10). An illustration of the dihedral angles (ϕ and ψ) of amino acid residues in a protein structure is called a Ramachandran plot. Plot regions assist verify and enhance the precision and caliber of three-dimensional protein representations by showing whether or not a protein structure is energetically advantageous. It is a crucial tool for comprehending how accurate protein conformations are structurally.
Allergenicity Analysis
Vaccine antigens can cause allergic reactions, resulting in severe complications. Revealing allergenicity of predicted peptides was an important consideration to avoid such complications. Both peptides were predicted as non-allergen by three methods, ML method, MERCI and Blast allergen search (Fig. 11).
Prediction of Transmembrane Topology
Proteins with multiple transmembrane (TM) spanning regions are difficult to clone, express and purify, therefore, antigens with <1 TM spanning region are considered suitable as vaccine candidates. DeepTMHMM server showed that there were no transmembrane regions in these peptides (Fig.12 (a) and Fig. 12(b)).
Molecular Docking
To analyze the interaction between the vaccine and the TLRs, molecular docking was used. HDOCK was used to predict the vaccine binding site by selecting the most thermodynamically favorable position. The vaccine construct was docked to TLR2, TLR3, TLR4, TLR7, TLR5 and TLR8. The server generated a binding complex with a docking score of −289.59, -296.78, -278.14, -278.28, -291.54 and -285.32 respectively. The minimum docking energy was of binding of the vaccine to TLR3 and therefore the most probable binding position. The ligand RMSD, calculated in angstroms, was determined to be 95.43. The final proposed docking positions of all TLRS and vaccine are displayed in Fig.13 (A-F).
Molecular Dynamics Simulation
The vaccine-MHC-II complex was simulated using WebGRO-UAMS to verify the root mean square
Fluctuations (RMSF) to further examine the stability of the structures. Most stable in silico vaccine designs have a greater RMSD value with similar fluctuation magnitudes and the value stabilizes after about 1ns. Simulation was done using force field GROMOS 43a1 at 300K temperature for 50 ns. All the residues except a few have fluctuations below 0.3 nm, which is generally the acceptable region (Fig.14).
Immune Infiltration Analysis
The clinical impact of different immune cells in NSCLC was studied. The abundance of immune cell types: PD-1 (PDCD1 gene), PD-L1 (encoded by CD274 gene) and CTLA-4 in the tumor microenvironment is estimated using a novel statistical method. Tumor purity represents the fraction of cancer cells in a tumor.
Correlation analysis of AZIN1 gene expression with three common immune checkpoints, PD-1, PD-L1 and CTLA-4, revealed that AZIN1 had positive correlation with the immune checkpoints (P value < 0.05, Fig. 15).
Immune Simulation
Using C-ImmSim Server, an immune simulation of the anticipated multi-epitope vaccination was carried out. IgM + IgG and IgM antibodies were detected in the initial immunization, according to the immunological simulation data (Fig. 16A). Due to the generation of total B-lymphocytes, the antigen count reduced as the antibody level in the immune response increased (Fig. 16B). During the immunological response, active TH-cell populations (Fig.16C) also rose in addition to B-cells. In the three phases following immunization, the level of interferon gamma (IFN-γ) had grown (Fig. 16E), and the active TC-cell (cytotoxic T lymphocyte) count had demonstrated persistent expansion (Fig. 16D).
Immune response increases on day 5 of immunization. The memory TH cells increased after day 5 of immunization while antigen levels were reduced to almost zero by day 5. We had chosen time period of 30 days for this simulation and antibody titer was seen during this entire period. IFN-γ levels were highly elevated, pointing towards the activation of apoptotic cell death of cancer cells. Simpson index (16D) indicated high diversity of interleukin-2 (IL-2) following immunization (Fig. 16E). It ranges from 0-1, with 0 representing infinite diversity and 1 representing no diversity at all. Low diversity of IL-2 is associated with autoimmune diseases and vaccine inhibiting immune response. These results revealed that our vaccine can induce a strong immune response.
Codon Optimization of the Vaccine Construct
The vaccine was subjected to reverse translation as a final step, producing a 150-nucleotide cDNA sequence. With JCat, the user can limit the use of restriction enzymes and stop them from interacting with particular cleavage sites. A cDNA sequence and the matching Codon Adaption Index (CAI) values are both returned by the algorithm. CAI values range from 0 to 1; value of 1 indicates ideal codon.
The codon was adjusted in relation to the mechanism of Homo sapiens gene expression using JCat. For comparison, an optimal GC-content proportion is between 30 and 70%, and a codon adaptation index (CAI) value is larger than 0.80. Gene expression is likely to be successful within these ranges. It can be concluded that the sequence might be integrated into the human genome because a GC-content proportion of 65.45% and a CAI value of 0.959 were computed (Fig.17).
Discussion
Immunotherapy is emerging as a popular and effective strategy in cancer treatment. Cancer treatment mostly depends on T-cell checkpoint regulation and/or antigen-specific immune responses [37]. According to Khatami et al. (2020), bioinformatics has altered the way the information is shared in the domains of biology and medicine and created new opportunities for the development of innovative molecules and procedures [38].
As far as vaccines for NSCLC are concerned, Cimavax and Tecemotide vaccines that target epidermal growth factor receptor (EGFR) and MUC1, respectively are not specific to NSCLC as these act on increased levels of the circulating markers of cancer, however, these are not the important drivers of lung cancer. Moreover, none of these alone have been found to improve the overall survival to a significant level and are being used in combination with other therapies [39]. AZIN1 on the other hand, has been identified as a potential target for NSCLC. It inhibits the degradation of DeltaNp73 functioning as an apoptosis suppressor when exposed to genotoxic stress [40]. It has been demonstrated that AZIN1 regulates tumor behavior by modulating centrosome duplication and upregulating loricrin to promote differentiation of cancer cells. AZIN1 RNA editing and its involvement in tumorigenesis of non-small-cell lung cancer has been confirmed by previous studies. Editing results in change of serine to glycine at residue 367 of AZIN1. This edited form is more stable and has more affinity to antizyme than wild type AZIN1. Suppressing AZIN1 editing has contributed to the development of novel RNA therapies for cancer treatment [41, 37]. AZIN1 is a novel driver of metastatic progression in different cancers and plays role in migration of cancer cells [42]. Our previous study also proved that AZIN1 centric DNB may play role at stage 2A and 2B making it the tipping point of metastasis in NSCLC [15]. AZIN1 is over-expressed in NSCLC making it a tumor associated antigen. AZIN1 expression also shows a significant positive correlation with immune checkpoints PD-1, PD-L1, and CTLA-4, suggesting that high AZIN1 expression contributes to an immunosuppressive tumor microenvironment [43, 44].
All these studies, therefore, strongly support that a multi-epitope vaccine predicted in our study targeting a novel tumor-associated antigen AZIN1 serves as a specific marker for NSCLC unlike other targets mentioned above thereby reducing the possibility of immune escape or development of resistance to the target.
Many studies have already shown the proven efficacy of peptide based multi-epitope vaccine in many cancers [45]. Potential advantages of such epitope-based vaccines include increased safety, the opportunity to rationally engineer the epitopes for increased potency and effect, and the ability to focus immune responses on conserved epitopes [46]. Vaccine construct is a long peptide incorporating multiple epitopes for both MHC-I and MHC-II makes it a good choice as immunization candidate as reported in many researches [47, 48]. A positive correlation between expression of AZIN1 and the degree of infiltration of PD-1, PD-L1 and CTLA-4 cells suggested that AZIN1 may have role in the regulation of the tumor immune microenvironment.
Furthermore, to estimate the feasibility of vaccine in the wet lab, the instability index (II) was calculated, which came out to be 10.12 revealing vaccine is stable in labs. Vaccines have to travel through the body. For efficient transportation into the body, strong binding affinity of vaccine with Toll-like receptors (TLRs) is needed. Docking studies revealed stable vaccine-TLR3 complexes. Studies indicate interesting and opposing role of TLR3 in lung cancer depending on its expression on tumour verses immune cells. Its increased expression in tumour cells is associated with direct apoptosis of tumor cells by activating lung dendritic cells to elicit positive immune responses resulting in better outcomes in early stage lung cancer in smokers. On the other hand, its presence in immune cells primarily macrophages, elicited poor prognosis in patients’ survival [49, 50]. Even our previous study showed that AZIN1 acts as tipping point of metastasis between stages 2A-2B.
Thus, it can be concluded that the predicted vaccine could be used as a therapeutic vaccine at the early stages of cancer for effective results. However, the present study has limitations because of use of only in silico models. In vitro or in vivo experiments are crucial for filling and bridging the gap with an in-silico experiment. Several in vitro experiments need to be performed such as cloning for recombinant protein expression, ELISA for analysing the interferons, antibodies, as well as cytokines when they interact with our designed vaccine regarding lung cancer mitigation. Once proved, an in vivo study (rat/mice) should be conducted for further validation for example, injecting the constructed vaccine in rat/mice models and observing their expression and its effect. Further, a flow cytometry assay could be used to analyse and count the number of memory cells, cytotoxic and helper T cells.
Acknowledgements
Authors wish to acknowledge Indira Gandhi National Open University, New Delhi for providing all the necessary facilities and research fellowship to AC.
Conflict of Interest
The authors declare that they have no competing financial interests.
- Siegel RL, Miller KD, Jemal A (2019) Cancer statistics, 2019. CA a Cancer Journal for Clinicians [Internet]. 69: 7-34.
- Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, et al. (2013) Predictive gene signature in MAGE-A3 Antigen-Specific Cancer immunotherapy. Journal of Clinical Oncology [Internet]. 31: 2388-95.
- Majeed U, Manochakian R, Zhao Y, Lou Y (2021) Targeted therapy in advanced non-small cell lung cancer: current advances and future trends. Journal of Hematology & Oncology [Internet]. 14.
- Korpanty GJ, Graham DM, Vincent MD, Leighl NB (2014) Biomarkers That Currently Affect Clinical Practice in Lung Cancer: EGFR, ALK, MET, ROS-1, and KRAS. Front Oncol. 4: 204.
- Peng L, Li H, Yan W, Liu Y, Zhu H and Hong Y (2025) Integrative profiling of lung cancer biomarkers EGFR, ALK, KRAS, and PD-1 with emphasis on nanomaterials-assisted immunomodulation and targeted therapy. Front. Immunol. 16: 1649445.
- Rother C, John T, Wong A (2024) Biomarkers for immunotherapy resistance in non-small cell lung cancer. Front. Oncol. 14: 1489977.
- Xue H, Fan Y, Li Y, Zhao Q, Zhang X, et al. (2025) Tumor-infiltrating lymphocytes in NSCLC: from immune surveillance to immunotherapy. Front Immunol. 16: 1610998.
- Wu J, Lin Z (2022) Non-Small cell lung cancer Targeted therapy: Drugs and mechanisms of drug resistance. International Journal of Molecular Sciences [Internet]. 23:15056.
- Wang X, Ward PA (2012) Opportunities and challenges of disease biomarkers: a new section in the journal of translational medicine. Journal of Translational Medicine, 10.
- Hoang PH, Landi MT (2022) DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers [Internet]. 14: 961.
- Giaccone G (2002) Targeted therapy in non-small cell lung cancer. Lung Cancer [Internet], 38:29-32.
- Tulluri V, Nemmara VV (2021) Role of antizyme inhibitor proteins in cancers and beyond. OncoTargets and Therapy [Internet], 14: 667-682.
- Olsen RR, Zetter BR (2011) Evidence of a role for antizyme and antizyme inhibitor as regulators of human cancer. Molecular Cancer Research [Internet]. 9: 1285-93.
- Hu X, Chen J, Shi X, Feng F, Lau KW, et al. (2017) RNA editing of AZIN1 induces the malignant progression of non-small-cell lung cancers. Tumor Biology [Internet]. 39: 101042831770000.
- Chauhan A, Kalra K, Kalra S (2024) AZIN1-centric dynamic network biomarker of tipping point in non-small cell lung cancer metastasis. Journal of Applied Biology & Biotechnology [Internet].
- Jespersen MC, Peters B, Nielsen M, Marcatili P (2017) BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Research [Internet]. 45: W24-W29.
- Chou PY, Fasman GD (1979) Prediction of the Secondary Structure of Proteins from their Amino Acid Sequence. Advances in Enzymology and Related Areas of Molecular Biology/Advances in Enzymology and Related Subjects [Internet]. 45-148.
- Emini EA, Hughes JV, Perlow DS, Boger J (1985) Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. Journal of Virology [Internet]. 55: 836-39.
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, et al. (2011) scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology [Internet] 7.
- Kolaskar AS, Tongaonkar PC (1990) A semi‐empirical method for prediction of antigenic determinants on protein antigens. FEBS Letters [Internet]. 2760: 172-4.
- Parker JMR, Guo D, Hodges RS (1986) New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and x-ray-derived accessible sites. Biochemistry [Internet]. 25: 5425-32.
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, et al. (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology [Internet] 7.
- Ghaffari-Nazari H, Tavakkol-Afshari J, Jaafari MR, Tahaghoghi-Hajghorbani S, Masoumi E, et al. (2015) Improving Multi-Epitope Long Peptide Vaccine Potency by Using a Strategy that Enhances CD4+ T Help in BALB/c Mice. PLoS ONE [Internet], 10: e0142563.
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, et al. (2003) Protein identification and analysis tools in the ExPASY server. Humana Press eBooks [Internet] 531-552.
- Saha S, Raghava GPS (2006) AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Research [Internet]. W202-W209.
- Schwede T (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Research [Internet] 31: 3381-85.
- Alugoju P (2023) Protocol 2: MD Simulation with Gromacs V1.
- Galon J, Bruni D (2019) Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nature Reviews Drug Discovery [Internet] 18:197-218.
- Zhao Y, Schaafsma E, Gorlov IP, Hernando E, Thomas NE, et al. (2018) A leukocyte infiltration score defined by a gene signature predicts melanoma patient prognosis. Molecular Cancer Research [Internet], 17: 109-119.
- Li T, Fu J, Zeng Z, Cohen D, Li J, et al. (2020) TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Research [Internet]. 48: W509-14.
- Rapin N, Lund O, Bernaschi M, Castiglione F (2010) Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE [Internet]. 5: e9862.
- Bhatnager R, Bhasin M, Arora J, Dang AS (2020) Epitope based peptide vaccine against SARS-COV2: an immune-informatics approach. Journal of Biomolecular Structure and Dynamics [Internet] 39: 5690-705.
- Mauro VP (2018) Codon optimization in the production of recombinant biotherapeutics: Potential risks and considerations. BioDrugs [Internet]. 32: 69-81.
- Grote A, Hiller K, Scheer M, Munch R, Nortemann B, et al. (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Research [Internet]. 33(Web Server): W526-W531.
- Solanki V, Tiwari M, Tiwari V (2019) Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Scientific Reports [Internet], 9.
- Wu CY, Monie A, Pang X, Hung CF, Wu Tc (2010) Improving therapeutic HPV peptide-based vaccine potency by enhancing CD4+ T help and dendritic cell activation. Journal of Biomedical Science [Internet]. 17: 88.
- Suckow MA (2013) Cancer vaccines: Harnessing the potential of anti-tumor immunity. The Veterinary Journal [Internet]. 198: 28-33.
- Khatami SH, Taheri-Anganeh M, Arianfar F, Savardashtaki A, Sarkari B, et al. (2020) Analyzing Signal Peptides for Secretory Production of Recombinant Diagnostic Antigen B8/1 from Echinococcus granulosus: An In silico Approach. PubMed [Internet]. 9: 1-10.
- Flores Banda JS, Gangane S, Raza F, Massarelli E (2025) Current Development of Therapeutic Vaccines in Lung Cancer. Vaccines (Basel). 13: 185.
- Dulloo I, Gopalan G, Melino G, Sabapathy K (2010) The antiapoptotic DeltaNp73 is degraded in a c-Jun–dependent manner upon genotoxic stress through the antizyme-mediated pathway. Proceedings of the National Academy of Sciences [Internet]. 107: 4902-07.
- Hu Y, Wei X, Lv Y, Xie X, Yang L, et al. (2020) EIF3H interacts with PDCD4 enhancing lung adenocarcinoma cell metastasis. PubMed [Internet]. 10: 179-195.
- Ghalali A, Wang L, Stopsack KH, Rice JM, Wu S, et al. (2022) AZIN1 RNA editing alters protein interactions, leading to nuclear translocation and worse outcomes in prostate cancer. Experimental & Molecular Medicine [Internet]. 54: 1713-26.
- Hu X, Chen J, Shi X, Feng F, Lau KW, et al. (2017) RNA editing of AZIN1 induces the malignant progression of non-small-cell lung cancers. Tumour Biol. 39: 1010428317700001.
- Cai Y, Wu Q, Liu Y, Wang J (2020) AZIN1-AS1, A Novel Oncogenic LncRNA, Promotes the Progression of Non-Small Cell Lung Cancer by Regulating MiR-513b-5p and DUSP11. Onco Targets Ther. 13: 9667-78.
- Sanami S, Azadegan-Dehkordi F, Rafieian-Kopaei M, Salehi M, Ghasemi-Dehnoo M, et al. (2021) Design of a multi-epitope vaccine against cervical cancer using immunoinformatics approaches. Scientific Reports [Internet]. 11.
- Zhou WY, Shi Y, Wu C, Zhang WJ, Mao XH, et al. (2009) Therapeutic efficacy of a multi-epitope vaccine against Helicobacter pylori infection in BALB/c mice model. Vaccine [Internet]. 27: 5013-19.
- Feyerabend S, Stevanovic S, Gouttefangeas C, Wernet D, Hennenlotter J, et al. (2009) Novel multi‐peptide vaccination in Hla‐A2+ hormone sensitive patients with biochemical relapse of prostate cancer. The Prostate [Internet]. 69: 917-27.
- Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, et al. (2012) Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nature Medicine [Internet]. 18: 1254-61.
- Bianchi F, Milione M, Casalini P, Centonze G, Le Noci VM, et al. (2019) Toll-like receptor 3 as a new marker to detect high risk early stage non-Small-Cell lung cancer patients. Sci Rep, 9: 14288.
- Bianchi F, Alexiadis S, Camisaschi C, Truini M, Centonze G, et al. (2020) TLR3 expression induces apoptosis in human non-Small-Cell lung cancer. Int J Mol Sci, 21: 1-14.
FIGURE 1
Figure 1: Flowchart for the Methodology for Prediction of Multiepitope Vaccine
FIGURE 2
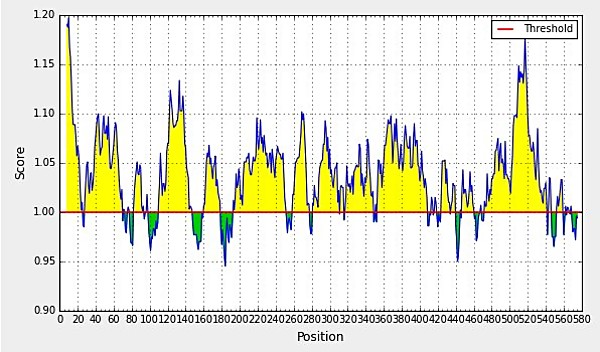
Figure 2A: BepiPred linear epitope prediction; yellow areas above the threshold (red line) are proposed B cell epitopes, and the green areas are not. [Threshold value=1.00]. Yellow color indicates the antigenic nature of the residues
FIGURE 3
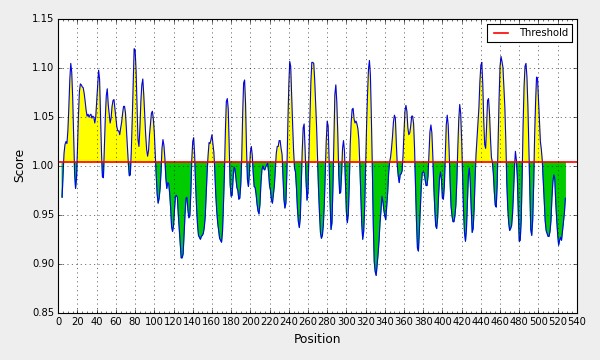
Figure 2B: Chau Fasman Prediction of Turns
FIGURE 4

Figure 2c: Karplus Method Predicted Chain Flexibility in Proteins
FIGURE 5

Figure 2D: Kolaskar and Tongaonkar Antigenicity Prediction
FIGURE 6

Figure 2E: Emini Surface Accessibility Prediction; Yellow Areas above the Threshold (Red Line) are Proposed to be Incorporated in B Cell Epitopes, and the Green Areas Are Not
FIGURE 7

Figure 2F: Parker Hydrophilicity Prediction; Yellow Areas above the Threshold (Red Line) Will Have Properties to Be B cell Epitopes
FIGURE 8

Figure 3: Vexigen Score of the Selected B-Cell Epitopes
FIGURE 9

Figure 4: Details of MHC I Epitopes
FIGURE 10
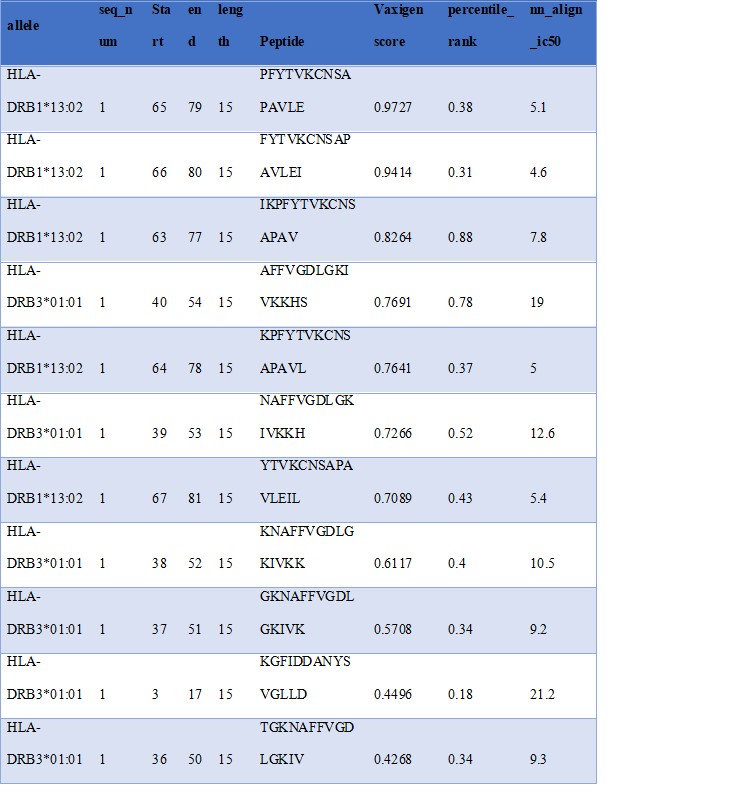
Figure 5: Details of the Selected MHC II Epitopes
FIGURE 11

Figure 6: Overlapping B-cell epitopes and T-cell epitopes. First immunogenic peptide comprise of overlapping B1, T9, T12 and T14 epitopes whereas second has overlapping T15, T6 and B3 epitopes
FIGURE 12
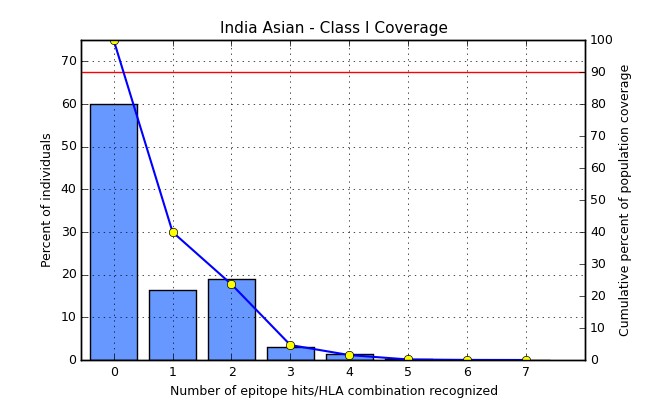
Figure 7: Population Coverage Analysis: Prediction of the population coverage for three potential vaccine candidates with MHC Class I HLA alleles around the Asian continent
FIGURE 13
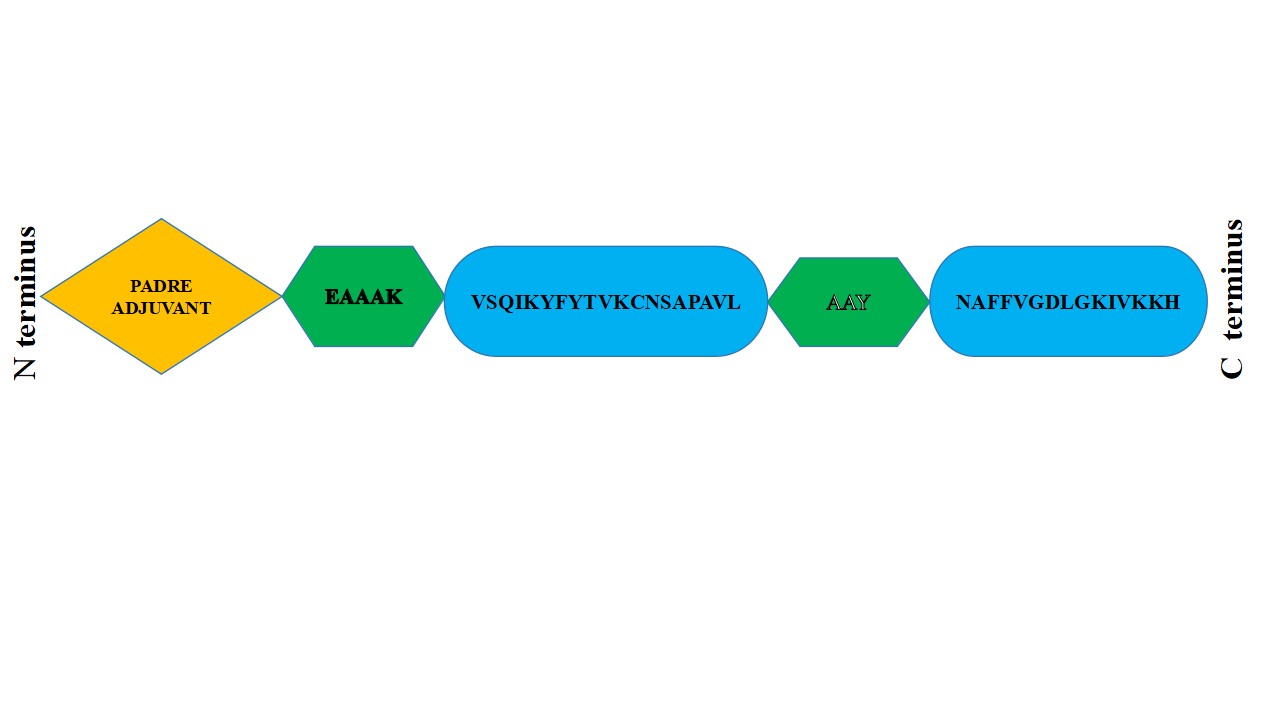
Figure 8: Schematic representation of vaccine construct: an adjuvant (yellow) linked by EAAAK linker (green) at the N-terminal end. AAY linkers (green) connect epitopes
FIGURE 14

Figure 9: Tertiary Structure Model of Vaccine Construct
FIGURE 15
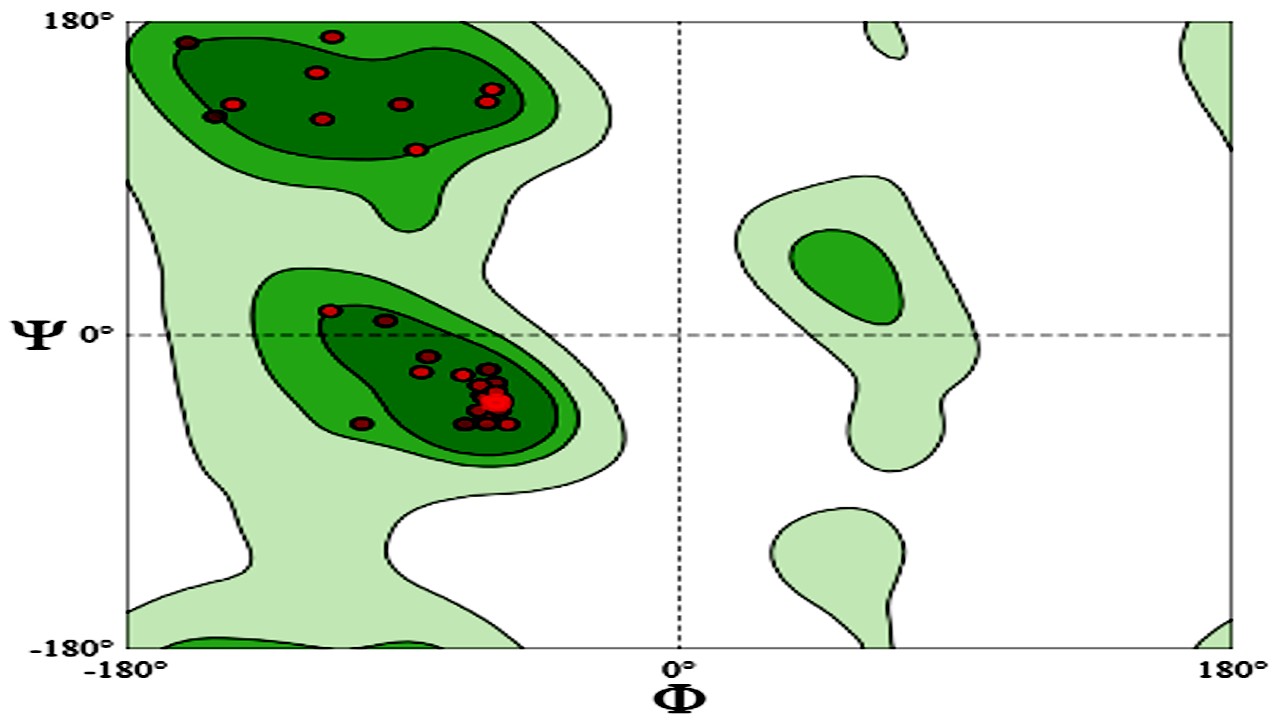
Figure 10: Ramachandran Plot of the Vaccine Construct
FIGURE 16
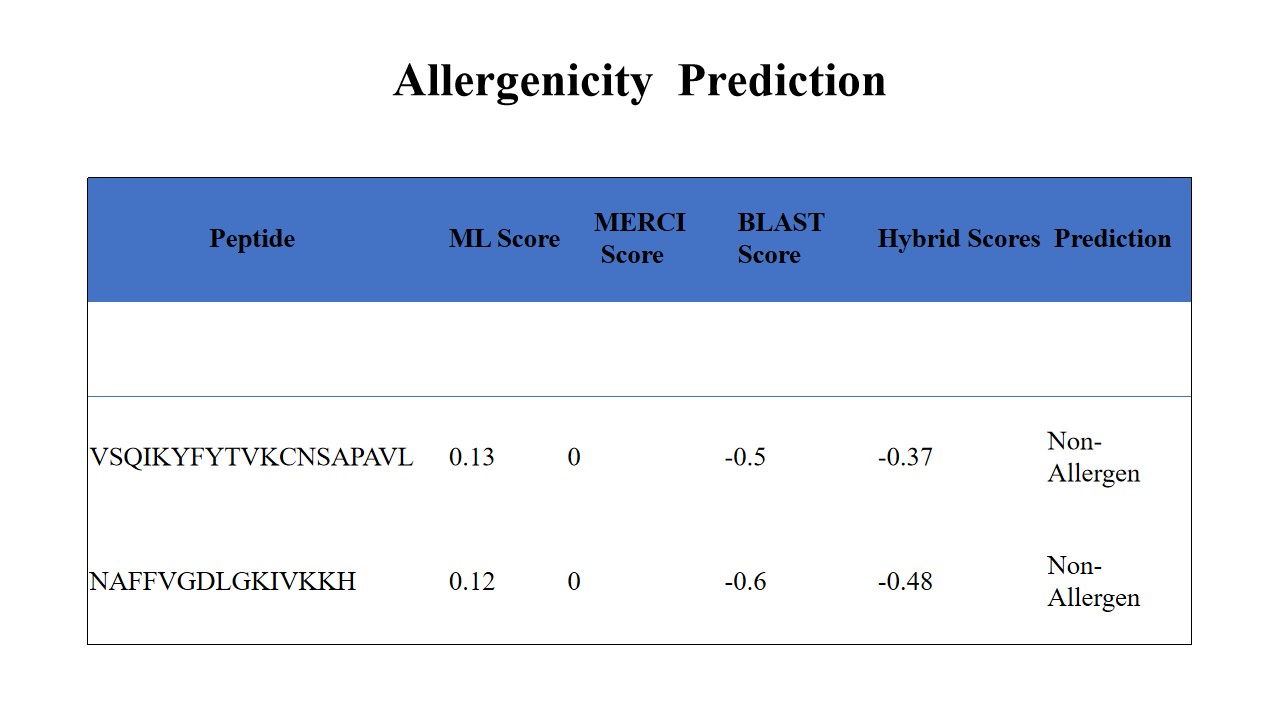
Figure 11: Allergenicity Prediction
FIGURE 17
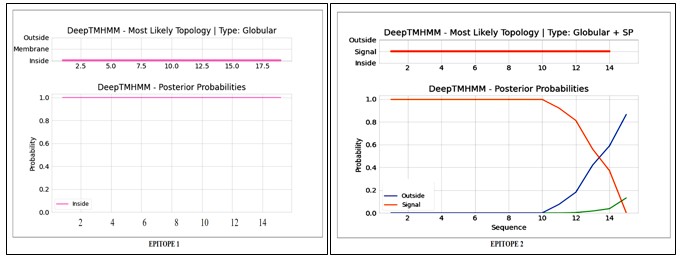
Figure 12(A): Topology Prediction of Epitope 1; 12(B): Topology Prediction of Epitope 2
FIGURE 18
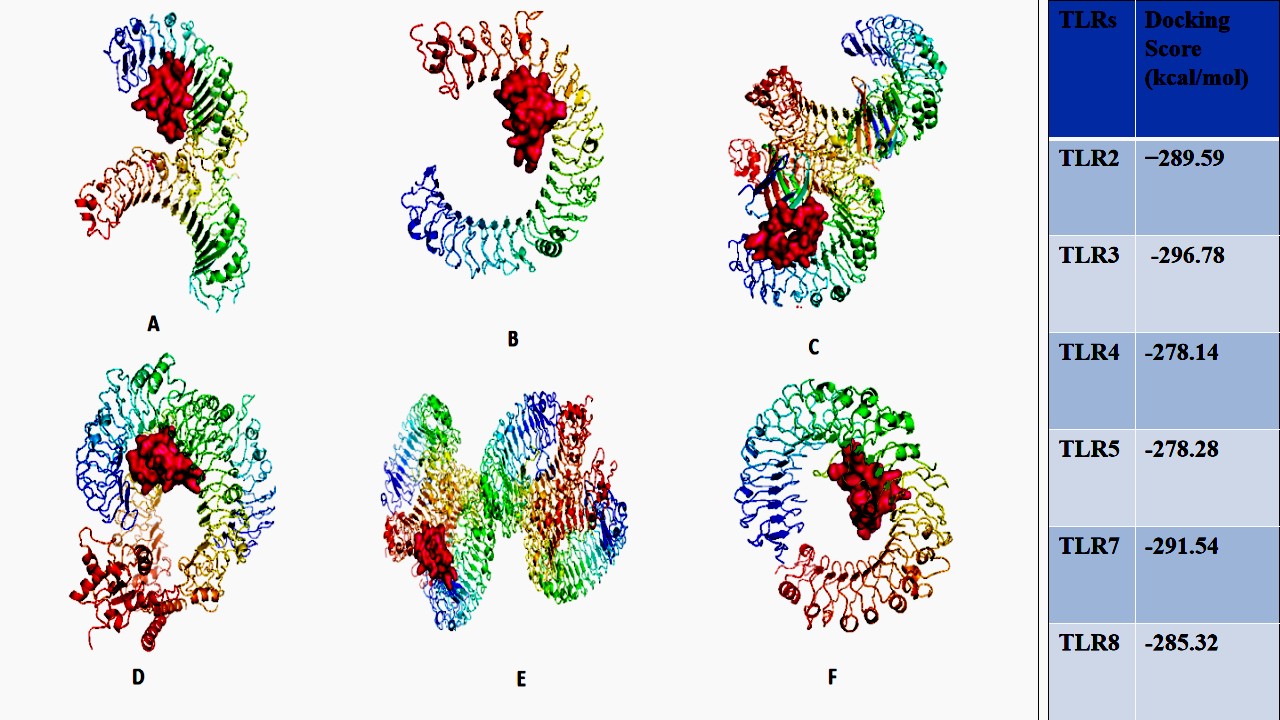
Figure 13: Vaccine construct docked to TLRs. TLR2, TLR3, TLR4, TLR5, TLR7 and TLR8(A-F)
FIGURE 19
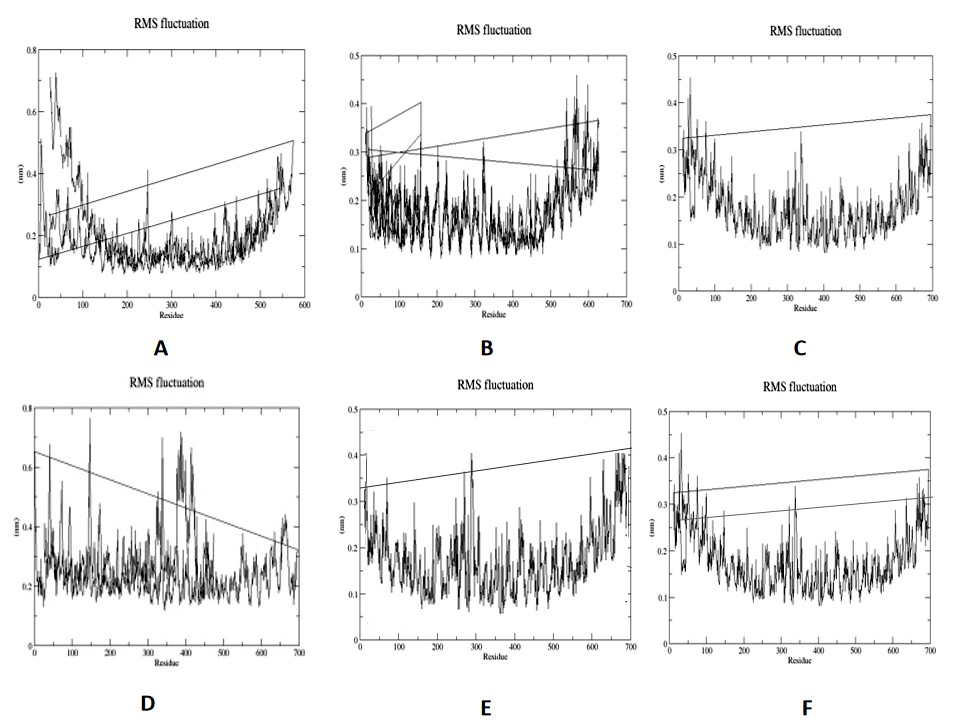
Figure 14: Root Mean Square fluctuations of vaccine-TLR2 complex(A) ,vaccine-TLR3 complex(B) ,vaccine-TLR4 complex(C) ,vaccine-TLR5 complex(D) ,vaccine-TLR7 complex(E) ,vaccine-TLR8 complex(F) using forcefield GROMOS 43a1 at 300K temperature for 50 ns
FIGURE 20
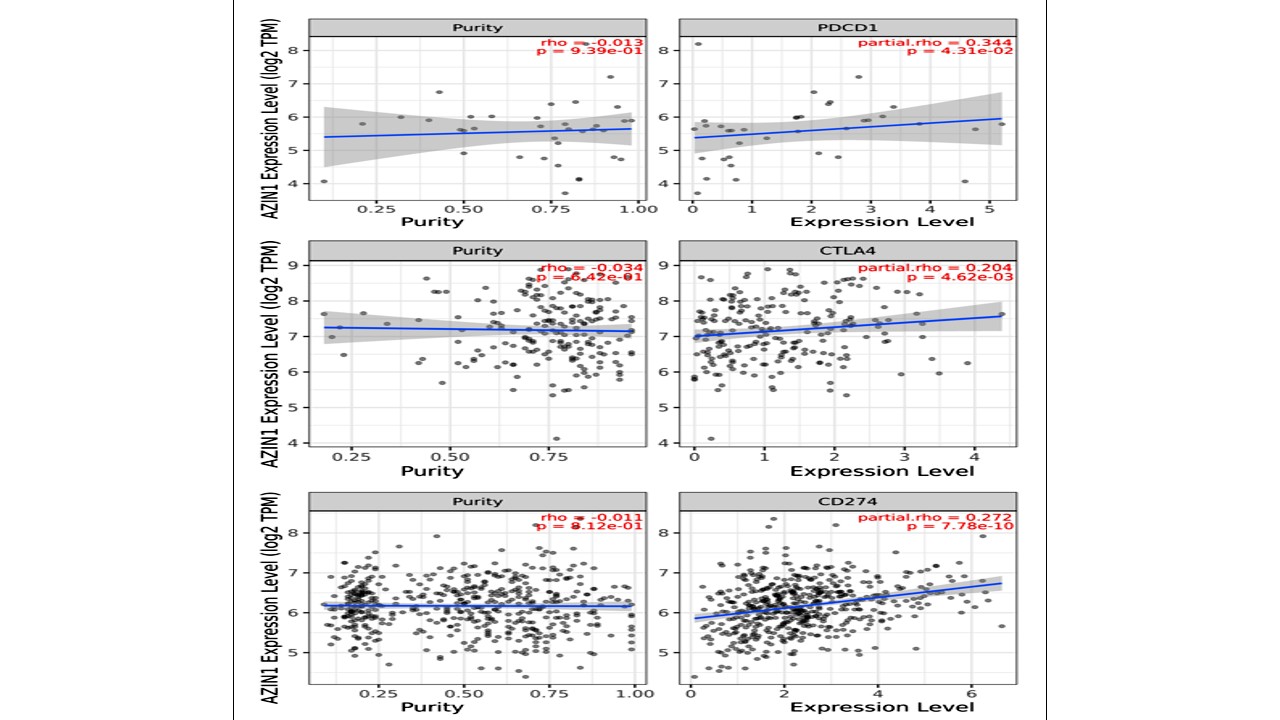
Figure 15: TIMER 2.0 scatter plot showing Spearman correlation between the expression of AZIN1 and immune checkpoints of PD-1, PD-L1 and CTLA-4.Tumor purity (left) and filtration level (right)
FIGURE 21
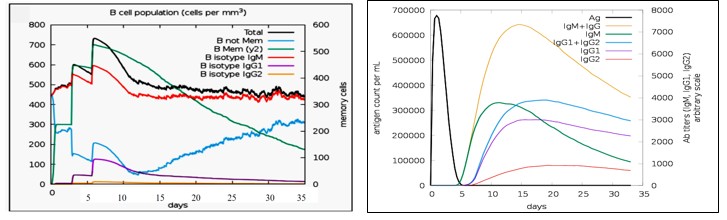
Figure 16(A): The Immune Simulation Using C-Immsim. Production Of B-Cell Populations Which Could Produce Antibodies of Various Subtypes After Vaccination. 16(B): The Production of Antibodies Representing Activation of the Immune Response after Vaccination. Various Subtypes of Immunoglobulins are Shown in Different Colors
FIGURE 22
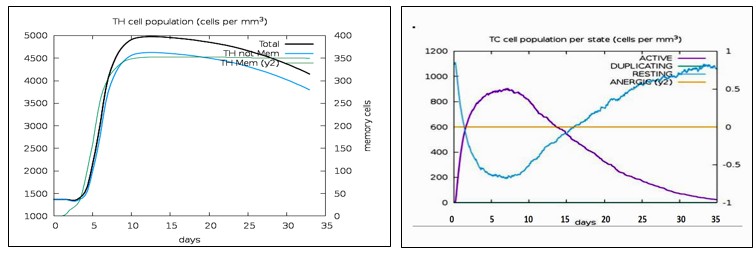
Figure 16(C): The generation of memory and non-memory HTL populations after vaccination. Fig 16(D): The CTL populations in various states. The RESTING indicates to the cells, which were not shown to the antigens while ANERGIC indicates the tolerance level of antigen.
FIGURE 23
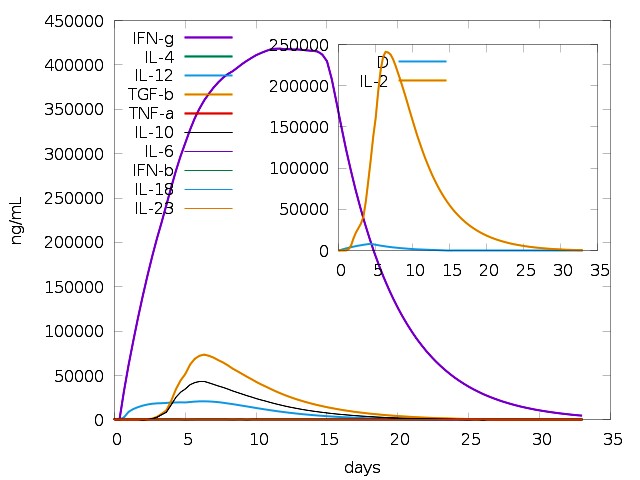
Figure 16(E): The Cytokine Profile Showed the Production of Various Cytokines After Vaccination.IFN-Γ Level Increased After Immunization. The Inset Graph Indicates the Simpson Index (D) Of IL-2
FIGURE 24
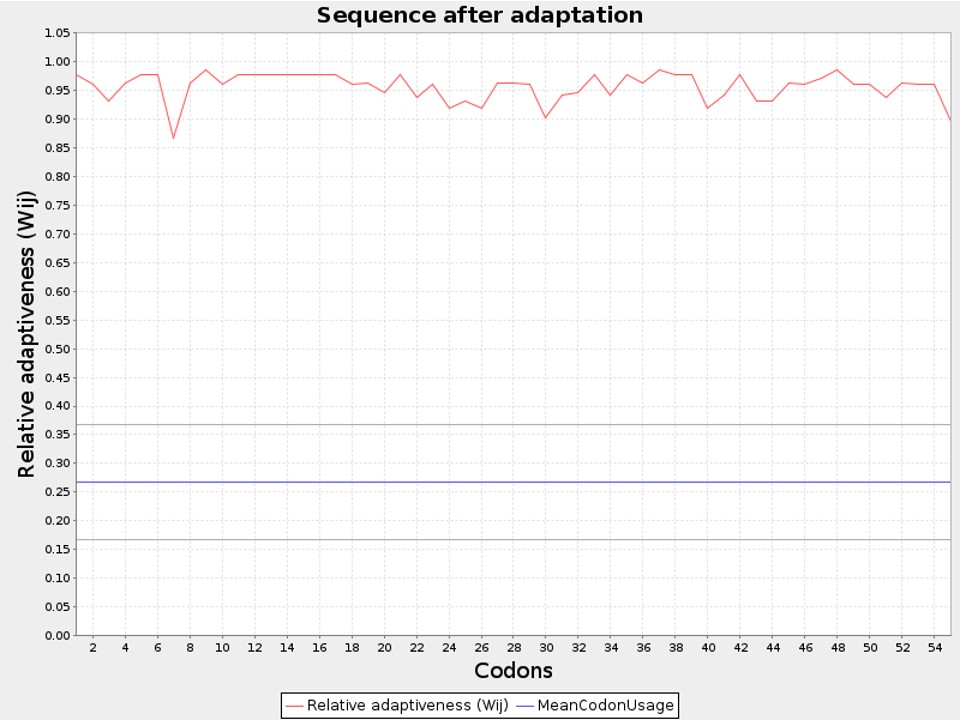
Figure 17: Codon Optimization Showing CAI Value of the Predicted Vaccine to be 0.959
Tables at a glance
Figures at a glance