Survival outcomes Associated with Various Resistance Mechanisms of Osimertinib in Chinese Advanced Non-Small Cell Lung Cancer Patients
Received Date: March 16, 2020 Accepted Date: April 07, 2020 Published Date: April 09, 2020
doi: 10.17303/jcrto.2020.8.204
Citation:Puyuan Xing (2020) Survival outcomes Associated with Various Resistance Mechanisms of Osimertinib in Chinese Advanced Non-Small Cell Lung Cancer Patients. J Cancer Res Therap Oncol 8: 1-10.
Abstract
Background:Various resistance mechanisms to osimertinib have been described; however, a comprehensive investigation of survival outcomes associated with these resistance mechanisms remains limited. Herein, we investigated the survival outcomes of osimertinib resistance mechanisms in Chinese non-small cell lung cancer (NSCLC) patients.
Methods: Targeted sequencing was performed on paired plasma samples collected prior to osimertinib and after the development of disease progression (PD) of 96 T790M-positive advanced NSCLC patients. The association between mutations acquired at PD and survival outcomes of the patients was also analyzed.
Results: Major acquired mutations were identified from our cohort including 25% EGFR, predominantly C797 and L792, 16% MET amplification, 8% TP53, 4% KRAS, 4% RET fusions, 4% ERBB2 amplification and 6.25% RB1. At baseline, 61 and 35 patients were EGFR exon 19 deletion (19del) and L858R mutants, respectively. Interestingly,19del-mutants acquired more mutations at PD(P=0.014), particularly in MAPK/PI3Kpathway (P=0.007) and TP53(P=0.021). On the other hand, acquired ERBB2 amplifications were only detected from L858R-mutants (P=0.047). Furthermore, 37.5% of the patients retained T790M while 62.5% lost T790M at PD. Our results revealed that patients retaining T790M, often associated with activation of bypass signaling pathways or continued EGFR activation through tertiary mutations, had a longer progression-free survival (PFS) (P=0.047) and overall survival (OS) (P=0.04) compared to patients with T790M loss, often with diverse and EGFR-independent mechanisms. Moreover, patients with acquired C797S had significantly longer PFS (P=0.031), while patients with acquired MET amplification had significantly shorter PFS (P=0.033).
Conclusion: Collectively, we revealed distinct survival outcomes associated with various resistance mechanisms, representing an important step in advancing the understanding of osimertinib resistance mechanisms.
Keywords: acquired resistance mechanism; osimertinib resistance; NSCLC; EGFR T790M.
Abbreviations: cfDNA, cell-free DNA; EGFR, epidermal growth factor receptor; NSCLC, non-small cell lung cancer; OS, overall survival; PD, disease progression; PFS, progression-free survival; TKI, tyrosine kinase inhibitor
Introduction
Osimertinib, a third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI), was developed to irreversibly bind to and inhibit mutated EGFR receptors [1-3]. Although it can target all mutated EGFR receptors, it is widely used in tumors that acquired EGFR T790M-mediated resistance from prior therapy with earlier generations of EGFR-TKI [1-3]. Patients with T790M-positive advanced non-small cell lung cancer (NSCLC) who progressed from prior EGFR-TKI have significantly longer median progression-free survival (PFS) on osimertinib compared with chemotherapy (10.1 vs. 4.4 months, P< 0.001) [3-4]. Despite remarkable clinical responses to osimertinib, patients still acquire resistance and develop disease progression between 10 to 19 months from initiation of osimertinib therapy [5,6]. Increasing efforts have been invested in elucidating the mechanisms of osimertinib resistance to develop novel strategies in overcoming this growing problem. Numerous studies have demonstrated the development of osimertinib resistance to be associated with EGFR-dependent mechanisms, predominantly through the acquisition of tertiary EGFR mutations including C797X, L792X, and L718X [7-9]. Moreover, a growing number of reports have also observed mutations in parallel or downstream pathways that mediate resistance to osimertinib including MET [8-13], ERBB2 [11,12], KRAS [8-10,12], BRAF [8,10], PIK3CA [8], and RET [8,14]. In addition, the histologic transformation from adenocarcinoma to small cell [8, 10, 15, 16] or squamous cell [10] has been documented as resistance mechanisms to osimertinib therapy, consistent with other generations of EGFR-TKI [17, 18]. Despite the availability of data on various molecular mechanisms of resistance to osimertinib [8-16], a comprehensive investigation of the clinical outcomes of patients with various resistance mechanisms remains unexplored. The information on clinical outcomes is critical in making therapeutic decisions for this subset of patients. In our study, we aimed to elucidate the osimertinib resistance mechanisms in EGFR T790M-positive Chinese NSCLC patients and assess their clinical outcomes based on their resistance mechanism.
Patients and Methods
Patients
A total of 96 EGFR T790M-positive advanced-stage NSCLC Chinese patients who progressed on prior first- or second-generation EGFR-TKI therapy and have detectable mutations at disease progression (PD) between October 2015 and January 2018 were enrolled in the study. Paired plasma samples were collected from the patients prior to the initiation of osimertinib therapy and after the development of PD. NSCLC was diagnosed according to the criteria by the 2015 World Health Organization histological classification of lung tumors [19]. Pathologic or clinical staging was according to the seventh edition of the American Joint Committee on Cancer [20]. Treatment responses were investigator-assessed based on Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 [21]. Medical records were retrieved to collect clinicopathologic data, treatment history and survival outcome. This study has been approved by the relevant Institutional Review Board of Cancer Hospital, Chinese Academy of Medical Science and performed in accordance with the standards set forth by the Declaration of Helsinki as revised in 2013. Written informed consent was provided by all the patients included in the study.
Cell-free DNA isolation and capture-based targeted DNA sequencing
As described previously [22], circulating cell-free DNA (cfDNA) was recovered from 4 to 5 ml of plasma using the QIAamp Circulating Nucleic Acid kit (Qiagen, Hilden, Germany). A minimum of 50 ng of cfDNA is required for NGS library construction. Fragments between 200 to 400base pairs (bp) from the cfDNA were end-repaired, phosphorylated and ligated with adaptors (Agencourt AMPure XP Kit, Beckman Coulter, CA, USA). Purified cfDNA with adaptors were then hybridized with capture probes baits, underwent hybrid selection with magnetic beads and PCR amplified. The quality and the size of the fragments were assessed using Qubit 2.0 fluorimeter with the dsDNA high-sensitivity assay kit (Life Technologies, Carlsbad, CA). Indexed samples were sequenced on Nextseq500 (Illumina, Inc., USA) with paired-end reads and average sequencing depth of 10,000X. A panel with 168 genes including 68 lung cancer-related genes and 100 other genes related to cancer development was used for targeted sequencing (Lung Plasma, Burning Rock Biotech, Guangzhou, China).
Sequence data analysis
Sequence data were mapped to the reference human genome (hg19) using Burrows-Wheeler Aligner v.0.7.10 [23]. Local alignment optimization and variant calling were performed using Genome Analysis Tool Kit v.3.2 [24], and VarScan v.2.4.3 [25]. Variants were filtered using the VarScan fpfilter pipeline, loci with depth less than 100 were filtered out. Base-calling in plasma samples required at least 8 supporting reads for single nucleotide variations (SNV) and 5 supporting reads for insertion-deletion variations (INDEL). Variants with population frequency over 0.1% in the ExAC, 1000 Genomes, dbSNP or ESP6500SI-V2 databases were grouped as single nucleotide polymorphisms (SNP) and excluded from further analysis. The remaining variants were annotated with ANNOVAR (2016-02-01 release) [26] and SnpEff v.3.6 [27]. Analysis of DNA translocation was performed using Factera v.1.4.3 [28]. Copy number variations (CNV) were analyzed based on the depth of coverage data of capture intervals using an in-house developed algorithm. The limit of detection for CNVs is 1.5 and 2.64 for deletions and amplification, respectively.
Statistical analysis
The differences in the groups were calculated and presented using either Fisher’s exact test or two-tailed Student’s t-test, as appropriate. Progression-free survival (PFS) was defined from the date osimertinib was administered until the evaluation of PD. Overall survival (OS) was defined from the date of diagnosis until the day of death or the last day of follow-up. PFS and OS curve was estimated using the Kaplan–Meier method and the differences among the groups were evaluated using the log-rank test. P-value with P< 0.05 was considered as statistically significant. All the data were analyzed using R statistics package (R version 3.4.0; R: The R-Project for Statistical Computing, Vienna, Austria).
Results
Patient characteristic
Our study cohort included a total of 96 patients, of which, 58% (56/96) were females and 42% (40/96) were males, with a median age of 56 years (ranging from 32 to 82 years). A majority were diagnosed with adenocarcinoma (96%, 92/96), while the remaining patients were diagnosed with adenosquamous carcinoma (2%, 2/96) and squamous cell carcinoma (2%, 2/96). All the patients were T790M-positive and had progressed from prior EGFR-TKI therapy. Table 1 summarizes the baseline clinicopathologic characteristics of the cohort.
Acquired mutations detected upon disease progression with osimertinib
To elucidate the mutations acquired after osimertinib therapy, capture-based targeted sequencing of the paired plasma samples collected prior to initiating osimertinib therapy (Figure 1A) and after developing resistance to osimertinib (Figure 1B) were performed using a 168-gene panel, spanning 0.273 mega bases of the human genome. To derive the acquired mutations at PD, the mutation profiles of each of the 96 patients from these two-time points were compared (Figure 1C).
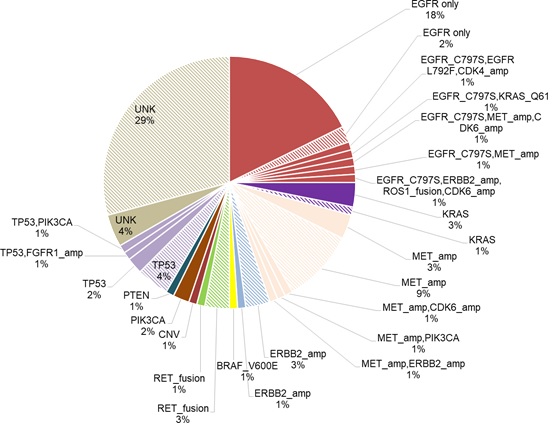
Among the patients in the cohort, a total of 258 acquired mutations spanning 90 genes were detected. Of which, 25% (24/96) of the patients acquired tertiary EGFR mutations, predominantly C797S (18/24). In addition, mutations in TP53 (8.3%, 8/96) and genes involved in bypass pathways including MET amplification (15.6%, 15/96), KRAS mutations (4%, 4/96), RET fusions (4%, 4/96) and ERBB2 amplification (3%, 3/96) were acquired by the patients at PD. In addition to the emergence of mutations at PD, 37.5% (36/96) of the patients retained EGFR T790M, while the remaining 62.5% (60/96) of the patients had lost the mutation at PD. Loss of EGFR T790M was defined as the detection of baseline EGFR sensitizing mutation (i.e. exon 19 deletions and L858R) from the sample with no concurrent detection of EGFR T790M at osimertinib PD. Figure 1 illustrates the major mutations acquired by the cohort at PD. According to this distribution, the major mutations known to mediate resistance to osimertinib acquired by this cohort at PD were EGFR tertiary mutations coupled with retention of EGFR T790M (18%), followed by the acquisition of MET amplification with loss of T790M (9%) and TP53 mutations with loss of T790M (4%). There were 32 (33%) of patients with no known resistance mechanisms at PD; among them, 4 patients (4%) retained EGFR T790M and the remaining 28 patients (29%) lost the mutation (Figure 2).
Our data further revealed that patients who retained T790M were likely to acquire tertiary EGFR mutations, including C797S/G, G796X, L718Q and L792F, and mutations in genes involved in bypass signaling pathways (P< 0.001, Figure 3). Interestingly, a majority of the patients with T790M retention who acquired the C797S/G at PD (n=19), acquired it on the same allele as the T790M (in cis configuration, C797S n=16, C797G n=1), while the remaining two patients acquired the C797S in trans to T790M. On the contrary, patients with T790M loss were likely to acquire diverse EGFR-independent resistance mechanisms (P< 0.001, Figure 3).
Acquired mutations based on baseline EGFR driver mutations
Next, we interrogated whether different baseline (prior to osimertinib treatment) EGFR driver mutations (EGFR 19 del vs L858R) are associated with certain osimertinib acquired mutations at PD. The cohort was further stratified according to baseline EGFR driver mutations. At baseline, 61 patients (63.5%, 61/96) harbored EGFR exon 19 deletion (19del)-mutants; while the remaining 35 (36.4%, 35/96) patients had EGFR L858R (Figure 3). At PD, all the patients with L858R (35/35) retained their baseline EGFR mutations. Meanwhile, 98.3% (60/61) of the 19del-mutants were detected with their baseline EGFR mutation with 1 patient who lost both EGFR19del and T790M and instead acquired an FGFR1 amplification and a TP53 E224D (Figure 2 and Figure S2). T790M were retained in 35.0% (21/60) and 42.9% (15/35) of the 19del-mutant and L858R-mutant patients, respectively. Tertiary EGFR mutations including C797S, C797G, G796X, and L718Q were detected from 16 and 8 patients with baseline 19del and L858R patients, respectively. Interestingly, G796R/S and L718Q were detected concurrently with C797G/S from 19del-mutant patients, while G796del and L718Q were detected exclusively from L858R-mutant patients. L792F concurrent to C797S was only detected from an L858R-mutant patient. Moreover, G724S were only detected from two 19del-mutant patients who lost EGFR T790M (Figure 3). However, no significant difference was found between patients with 19del and L858R in harboring tertiary mutations in G796X (P=1), C797X (P=0.589) and L718Q (P=0.299). Collectively, our study revealed patients with baseline EGFR 19del or L858R had comparable rates of T790M retention.
We identified that, on average, patients with baseline 19del had 142 mutations; in contrast, patients with L858R had 47 mutations. Patients with baseline 19del had significantly more mutations (P=0.014), particularly in MAPK/PI3K pathway (P=0.007, Figure 4A) and TP53 (P=0.021, Figure 3 and 4B) than patients with baseline L858R. MAPK/PI3K pathway-related mutations from literature as a resistance mechanism detected from 19del-mutants included BRAF, KRAS, PI3KCA, PTEN, RET, and MET, respectively (Figure 2). Interestingly, potentially actionable mutations including BRAF V600E, RET fusions, and MET amplification were mostly detected among 19del-mutant patients. However, they did not reach a significant difference due to a small cohort (BRAF V600E, P=1; RET, P=0.293; MET, P=0.4). Other mutations in the MAPK/PI3K pathway which were not established to be involved in acquired osimertinib or EGFR-TKI resistance were not significantly different between 19del- (n=33) and L858R- (n=29) mutant patients (P=0.075, Figure 4A). Furthermore, acquired ERBB2 amplifications were only detected from 3 L858R-mutant patients (P=0.045, Figure 4C). Collectively, our study revealed a distinct mutation landscape at PD between patients with EGFR 19del or L858R at baseline, suggesting their heterogeneity of resistance mechanisms to osimertinib.
Survival outcomes
Next, medical records were retrieved to assess their PFS and OS. Analyses revealed that patients with T790M loss (n=60) had significantly shorter PFS (median PFS 6.7 months vs. 10.4 months, P=0.047, Figure 5A) and OS (median OS 15.7 months vs. 17.5 months, P=0.04, Figure 5B) compared to patients who retained T790M at PD. Furthermore, we also revealed that patients with acquired tertiary EGFR mutations had significantly longer PFS (median PFS 10.5 months vs. 7 months, P=0.007, Figure 6A) and OS (median OS 18.1 months vs. 15.7 months, P=0.047, Figure 6B), particularly the PFS for patients who acquired EGFR C797X (median PFS 10.5 months vs. 7 months, P=0.021, Figure 6C). However, no significant difference in OS was observed for this particular tertiary mutation (median OS NA vs. 16.2 months, P=0.11, Figure 6D), potentially attributing to the immature OS of patients who acquired EGFR C797X. Furthermore, patients who acquired MET amplifications at PD had significantly shorter PFS (median PFS 5.3 months vs. 9.5 months, P=0.031, Figure 7A), but no significant difference in OS (median OS 13.4 months vs. 17.3 months, P=0.1, Figure 7B). Moreover, no significant difference was found between the PFS (P=0.407) and OS (P=0.657) of patients with baseline 19del and L858R.
Discussion
EGFR-TKIs have profoundly improved the prognosis of EGFR-mutant NSCLC patients; however, resistance inevitably arises in almost all patients with a median PFS under 12 months [5, 6]. Through the years, increasing efforts have been invested in elucidating the resistance mechanism and developing novel therapies to overcome EGFR-TKI resistance. Acquired resistance mechanisms to osimertinib have been investigated, revealing both EGFR-dependent and independent mechanisms [8-13]. However, information on the survival outcomes of the patients with diverse resistance mechanisms acquired from osimertinib therapy still remains limited, particularly in Chinese NSCLC patients. Herein, we investigated the acquired resistance mechanisms and survival outcomes of 96 Chinese osimertinib-treated advanced NSCLC patients who progressed from prior generations of EGFR-TKI.
Consistent with acquired mutations previously reported by numerous studies [8-13], the acquired mutations detected in our cohort were very heterogeneous. The major mutations acquired by our cohort at PD were tertiary EGFR mutations (25%, 24/96) and mutations in bypass pathways including MET amplification (15.6%, 15/96), KRAS mutations (4%, 4/96), RET fusions (4%, 4/96), and ERBB2 amplification (3%, 3/96). Moreover, our data also revealed that patients retaining T790M were likely to be associated with the activation of bypass signaling pathways or continued EGFR activation through tertiary mutations. On the contrary, patients with T790M loss acquired diverse and EGFR-independent resistance mechanisms. In our cohort, 62.5% (60/96) of the patients had EGFR T790M loss at PD, slightly higher than the previously reported rate of 47% [8]. Our finding of patients with T790M retention had longer PFS (P=0.005) and OS (P=0.037) was in agreement with other studies [10]. Moreover, we also observed that patients with acquired C797S had significantly longer PFS (P=0.031), while patients with acquired MET amplification had significantly shorter PFS (P=0.033). Taken together, our data suggest that the loss of T790M and the acquisition of mutations in genes involved in bypass pathway are associated with shorter PFS, which leads us to conclude that acquisition of EGFR-dependent resistance mechanisms are associated with better prognosis, while EGFR-independent resistance mechanisms are associated with a worse prognosis.
Moreover, we also analyzed whether patients harboring different EGFR sensitizing mutations at baseline would have distinct mutations at PD. In our cohort, all patients retained their EGFR sensitizing mutation at PD except for a 19del-mutant patient. Interestingly, 19del- and L858R-mutant patients, despite having similar treatment responses and survival outcomes to osimertinib, have differential mechanisms of acquired resistance. Our data revealed that 19del-mutants acquired more mutations at PD (P=0.014), particularly in MAPK/PI3K pathway (P=0.007) and TP53 (P=0.021), while acquired ERBB2 amplifications were only detected in L858R-mutants (P=0.047). To the best of our knowledge, no previous reports have described the distinct mechanisms acquired by 19del-mutants and L858R-mutants. Further studies with a larger cohort are required to establish these findings.
Numerous preclinical [29] and clinical studies [12,14] have demonstrated the efficacy of combination inhibitors targeting EGFR and bypass pathway-mediated resistance acquired during osimertinib therapy. Serial molecular profiling is necessary to guide health care providers and provide options for appropriate subsequent therapy for patients before or after treatment failure with osimertinib.
Conclusion
Collectively, our study revealed the distinct survival outcomes associated with various osimertinib resistance mechanisms, which could contribute to advancing our understanding of the mechanisms involved in osimertinib resistance. This study could also pave the way in developing novel therapeutic strategies to further improve the prognosis of patients who develop resistance to osimertinib.
Acknowledgments
The authors thank all the patients who participated in this study and their families. We also thank the investigators, study coordinators, operation staff, and the whole project team who worked on this study.
Funding
This study was supported by a grant from the Chinese Academy of Medical Science Initiative for Innovative Medicine (CAMS I2M) [grant number: 2017-I2M-2-003].
Author contributions
P. Xing, X. Hao, J. Li worked on the conception and design of the study. P. Xing, Y. Mu, S. Wang, and D. Ma collected the data. J. Lin assisted with the statistical analysis. H. Liu and X. Mao provided the methodology. P. Xing, Y. Mu, S. Wang, D. Ma, J. Lin, H. Liu, A. Lizaso, H. Han-Zhang, J. Xiang, and X. Mao analyzed the data. P. Xing, A. Lizaso, H. Han-Zhang wrote the manuscript in consultation with X. Hao and J. Li. All the authors approved the manuscript.
Conflict of interest
J. Lin, H. Liu, A. Lizaso, H. Han-Zhang, J. Xiang, and X. Mao are employees of Burning Rock Biotech. The other authors declare no potential conflicts of interest.
- Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. (2009) Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462: 1070-1074.
- Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. (2014) AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4:1046-1061.
- Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. (2015) AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 372: 1689-1699.
- Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. (2017) Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med. 376: 629-640.
- Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. (2018) Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med. 378: 113-125.
- Ahn MJ, Tsai CM, Shepherd FA, Bazhenova L, Sequist LV, Hida T, et al. (2019) Osimertinib in patients with T790M mutation-positive, advanced non-small cell lung cancer: Long-term follow-up from a pooled analysis of 2 phase 2 studies. Cancer. 125: 892-901.
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. (2015) Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat Med. 21: 560.
- Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. (2018) Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 4:1527-1534.
- Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. (2018) Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin Cancer Res. 24: 3097-3107.
- Lin CC, Shih JY, Yu CJ, Ho CC, Liao WY, Lee JH, et al. (2018) Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: a genomic study. Lancet Respir Med. 6:107-116.
- Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, et al. (2015) EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. 26: 2073-2078.
- Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L, et al. (2016) Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin Cancer Res. 22: 4837-4847.
- Ou SI, Agarwal N, Ali SM (2016) High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer. 98: 59-61.
- Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. (2018) Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 8:1529-1539.
- Kim TM, Song A, Kim DW, Kim S, Ahn YO, Keam B, et al. (2015) Mechanisms of Acquired Resistance to AZD9291: A Mutation-Selective, Irreversible EGFR Inhibitor. J Thorac Oncol. 10:1736-1744.
- Ham JS, Kim S, Kim HK, Byeon S, Sun JM, Lee SH, et al. (2016) Two Cases of Small Cell Lung Cancer Transformation from EGFR Mutant Adenocarcinoma During AZD9291 Treatment. J Thorac Oncol. 11:e1-4.
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 3: 75ra26.
- Shee-Chai C, Liam CK, Mun KS (2017) Small Cell Transformation and T790M Mutation as Coresistance Mechanisms for First-line Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor (TKI) Therapy Failure. J Thorac Oncol. 12: e171-e3.
- Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. (2015) The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol. 10: 1243-1260.
- Edge SB, Compton CC (2010) The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 17:1471-1474.
- Schwartz LH, Litiere S, de Vries E, Ford R, Gwyther S, Mandrekar S, et al. (2016) RECIST 1.1-Update and clarification: From the RECIST committee. European journal of cancer. 62: 132-137.
- Mao X, Zhang Z, Zheng X, Xie F, Duan F, Jiang L, et al. (2017) Capture-Based Targeted Ultradeep Sequencing in Paired Tissue and Plasma Samples Demonstrates Differential Subclonal ctDNA-Releasing Capability in Advanced Lung Cancer. J Thorac Oncol. 12: 663-672.
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25: 1754-1760.
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297-1303.
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22: 568-576.
- Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164.
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 6: 80-92.
- Newman AM, Bratman SV, Stehr H, Lee LJ, Liu CL, Diehn M, et al. (2014) FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics.30:3390-3393.
- Romaniello D, Mazzeo L, Mancini M, Marrocco I, Noronha A, Kreitman M, et al. (2018) A Combination of Approved Antibodies Overcomes Resistance of Lung Cancer to Osimertinib by Blocking Bypass Pathways. Clin Cancer Res. 24: 5610-5621.
FIGURE 1
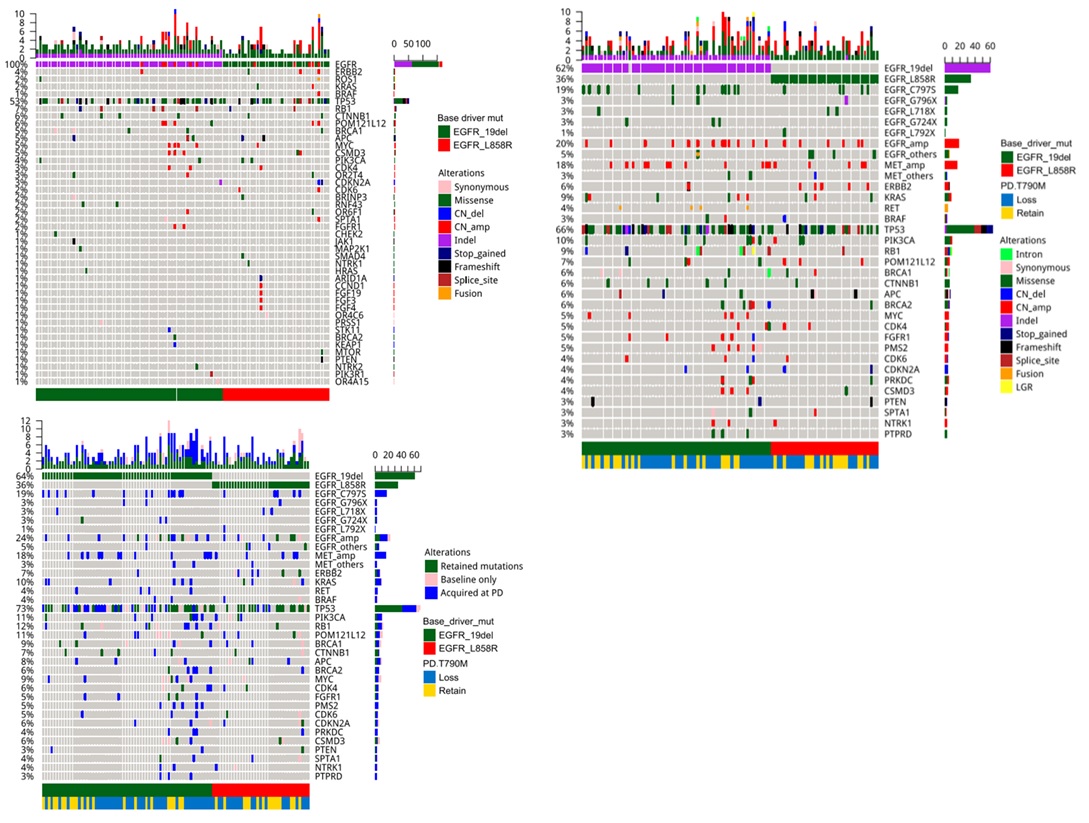
Figure 1:Mutation profile of the cohort prior to osimertinib therapy (A), and at disease progression with osimertinib (B). The mutation profiles from the two time-points were also compared and depicted in (C). A-C. The patients were grouped according to baseline EGFR mutations 19del (green) or L858R (red) as indicated by the bar located at the bottom of the oncoprint. B�C. An annotation depicting the EGFR T790M status of each patient at progression, whether lost (blue) or retained (yellow) was also added at the bottom of the oncoprint. Each column represents a patient and each row represents a gene. Table on the left represents the mutation rate of each gene. Top plot represents the overall number of mutations a patient carried. Different colors denote different types of mutation.
FIGURE 2
Figure 2: Distribution of the osimertinib-treated patients according to the major acquired mutations. Shaded slices refer to those with T790M loss.
FIGURE 3

Figure 3: Distinct acquired mutations between EGFR 19del and L858R-mutant patients. Summarized spectra of the mutations acquired by the cohort at PD. The first three rows reflect the baseline EGFR sensitizing mutations 19del and L858R and T790M and is the basis of the grouping of the patients. Each column represents a patient and each row represents a gene mutation indicated on the right. Percentage on the left and the histogram on the right represents the mutation rate of each gene.
FIGURE 4
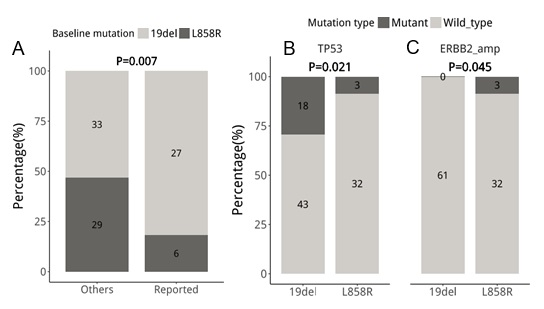
Figure 4: EGFR 19del-mutant patients acquired significantly more mutations in the MAPK/PI3K pathway (P=0.007) and TP53 (P=0.021), while ERBB2 amplifications were only detected from L858-mutant patients (P=0.045). Histograms illustrating the acquired mutations detected among 19del and L858R-mutant patients in A. MAPK/PI3K pathway; B. TP53 and C. ERBB2 amplification. Reported includes only specific mutations already established in the development of osimertinib or EGFR-TKI resistance, Others refers to mutations that have not been associated with the EGFR-TKI resistance mechanism.
FIGURE 5
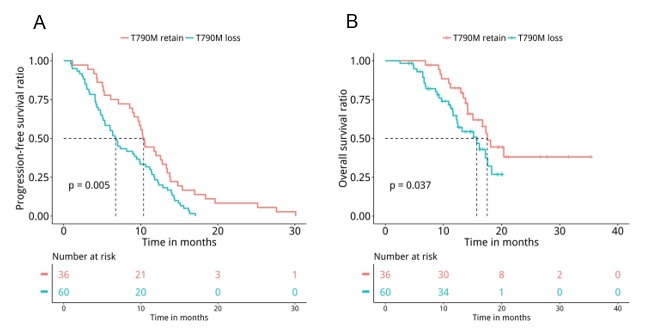
Figure 5: The loss of EGFR T790M at PD was associated with marginally shorter PFS (P=0.05) and significantly shorter OS (P=0.04). Kaplan-Meier analysis of the PFS and OS of the advanced NSCLC patients treated with osimertinib who retained (n=36) or lost (n=60) EGFR T790M at PD, indicated by red and blue lines, respectively. The risk table below illustrates the number of patients included per time point.
FIGURE 6
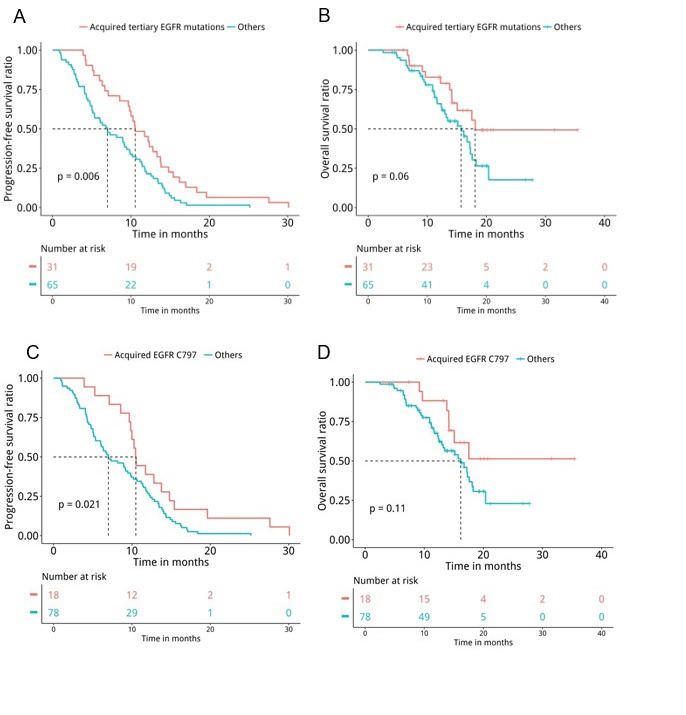
Figure 6:The acquisition of tertiary EGFR mutations, particularly C797X, at PD was associated with significantly longer PFS (P=0.006; C797 P=0.021) and a trend of longer OS (P=0.06). Kaplan-Meier analysis of the PFS and OS of the advanced NSCLC patients treated with osimertinib who acquired tertiary EGFR mutations (n=31) or other mutations (n=65) at PD, indicated by blue and red lines, respectively. The risk table below illustrates the number of patients included per time point.
FIGURE 7
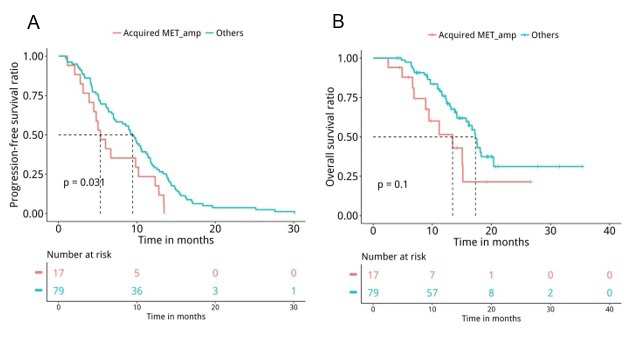
Figure 7:The acquisition of MET amplification at PD was associated with significantly shorter PFS (P=0.0321). Kaplan-Meier analysis of the PFS and OS of the advanced NSCLC patients treated with osimertinib who acquired MET amplification (n=17) or other mutations (n=79) at PD, indicated by blue and red lines, respectively. The risk table below illustrates the number of patients included per time point.
Tables at a glance
Figures at a glance