Abnormal Coronary Artery Origin in a Patient with Trisomy 13 (Patau Syndrome)
AFFILIATIONS
AFFILIATIONS
AFFILIATIONS
AFFILIATIONS
Received Date: March 13, 2025 Accepted Date: April 13, 2025 Published Date: April 16, 2025
doi: 10.17303/jcvm.2025.11.101
Citation: Yang Hu, Priya Upadhyay, Shiraz Maskatia, Jody E. Hooper, Darren Salmi (2025) Abnormal Coronary Artery Origin in a Patient with Trisomy 13 (Patau Syndrome). J Cardio Vasc Med 11: 1-7
Abstract
Trisomy 13 (Patau syndrome) is a rare genetic disorder that frequently involves nervous, orofacial, genitourinary, musculoskeletal, and cardiovascular organ systems. The syndrome carries a median survival of 7–10 days with less than 10% one-year survival. We present an autopsy of a 7-month-old male with trisomy 13 (karyotype: 47, XY, +13) who experienced postnatal complications such as necrotizing enterocolitis requiring bowel resection, refractory seizures, and respiratory failure necessitating tracheostomy but had otherwise no life-threatening complications in the weeks leading up to his death. On the day of his death, the patient was presumed to develop an arrhythmia, resulting in sudden cardiac death. Autopsy revealed a previously unreported coronary anomaly: a stenotic left coronary artery with high takeoff above the aortic commissure, accompanied by tetralogy of Fallot (VSD, overriding aorta, pulmonary stenosis, right ventricular hypertrophy) and patent foramen ovale. Pulmonary examination ruled out infection as the immediate cause of death. To our knowledge, this is the first report linking trisomy 13 to coronary ostial abnormalities, which may contribute to sudden cardiac death in this patient.
Keywords: Coronary Artery; Trisomy 13; Patau Syndrome; Pregnancies; Edward Syndrome
Introduction
Trisomy 13, or Patau syndrome, is a rare genetic disorder with an estimated prevalence of 1–2 cases per 10,000 pregnancies. This makes it about as common as trisomy 18, or Edward syndrome, but much less common than trisomy 21, or Down syndrome [1]. Due to high rates of stillbirth and elective termination, only 0.55 live births per 10,000 pregnancies are reported [2]. Among liveborn infants, nearly 50% die within the first week of life, and fewer than 10% survive beyond one year [2].
The earliest documented descriptions of trisomy 13 date to the 16th century, but Thomas Bartholin’s 1657 report of a patient with bilateral anophthalmos, facial anomalies, cleft lip, and polydactyly is widely recognized as the first definitive account [3]. The syndrome’s genetic basis was not elucidated until 1960, when Patau and colleagues identified an extra copy of chromosome 13 as the etiology of these congenital anomalies [4]. Their seminal case mirrored Bartholin’s findings while expanding the phenotypic spectrum to include hearing loss, cutaneous defects, seizures, developmental delay, and congenital heart disease [4].
Trisomy 13 is characterized by multisystem involvement, with frequent anomalies of the nervous, orofacial, genitourinary, musculoskeletal, and cardiovascular systems. Common manifestations include microcephaly, anophthalmia/microphthalmia, cleft lip/palate, urinary tract obstruction, polydactyly, and atrial or ventricular septal defects [5]. Congenital heart defects occur in approximately 80% of cases, with ventricular septal defects (VSD) seen in about half of the cases and atrial septal defects (ASD) seen in about 1/3 of the cases, though more complex lesions such as tetralogy of Fallot can be seen less frequently [5,6]. Notably, cardiac malformations rarely drive early mortality, which is instead attributed to central apnea, aspiration, hypoventilation, and upper airway insufficiency [6,7]. In fact, less than 10% of patients with trisomy 13 have a congenital heart defect that would be considered lethal [7].
Prognosis remains poor, with a median survival of 7–10 days [8]. While only 50% of infants survive the first week, only 6–12% reach one year of age [6,10-12]. This underscores the critical need for early recognition and tailored management strategies to address life-threatening complications.
Case Description
Clinical Course
The patient was a 7-month-old male with prenatally confirmed trisomy 13 (karyotype: 47, XY, +13, arr [13]x3). Prenatal ultrasound revealed double outlet right ventricle (DORV) with mild-to-moderate pulmonary annular hypoplasia, an enlarged hypoechoic right kidney, and a dysplastic left kidney. He was born at 38 weeks and 6 days gestation via induced vaginal delivery to a 39-year-old mother with an unremarkable medical history, within a non-consanguineous relationship.
Following delivery, the infant required positive pressure ventilation and prolonged neonatal intensive care unit (NICU) admission due to airway narrowing and congenital cardiac anomalies. At seven weeks of age, he developed Escherichia coli sepsis accompanied by pneumatosis intestinalis, consistent with necrotizing enterocolitis (NEC). Abdominal imaging demonstrated ascites suggestive of intestinal perforation, prompting emergency laparotomy with bowel resection and diverting ileostomy. Postoperatively, he experienced seizures managed with levetiracetam and phenobarbital. Persistent respiratory distress secondary to laryngomalacia and glossoptosis necessitated tracheostomy at one month of age with continuing ventilator dependence.
Prenatal echocardiography identified DORV without ductal dependency. Postnatal assessments revealed a total mixing lesion with target oxygen saturation (SpO2) of 75–85%, though SpO2 consistently ranged in the high 80s to low 90s. Serial echocardiograms demonstrated small-to-moderate anterior pericardial effusion, echogenic density on the atrial surface of the tricuspid valve, large perimembranous ventricular septal defect (VSD) with leftward septal bowing, mild-to-moderate systolic dysfunction of the left ventricle, and moderate hypertrophy with mild systolic impairment of the right ventricle. Despite a history of hypercyanotic (“tet”) spells, the patient remained hemodynamically stable.
At 7 months of age, he was discovered unresponsive and pulseless at home by his parents during the early morning hours. Immediate cardiopulmonary resuscitation (CPR) was initiated by the parents. Emergency Medical Services (EMS) arrived within 20 minutes, established intraosseous access, administered epinephrine, and continued advanced cardiac life support. The patient was airlifted to a tertiary care center within one hour, with ongoing resuscitative efforts during transport. Despite interventions by EMS, no return of spontaneous circulation (ROSC) was achieved. After approximately two hours of continuous efforts, the patient was pronounced deceased.
Autopsy
A complete autopsy was performed with findings summarized in Table 1. Examination of the heart revealed a stenotic left coronary ostium with an abnormally high takeoff and aberrant origin above the commissure of the left and posterior aortic cusps (Figure 1). The right coronary ostium had normal configuration. Cardiac examination also showed tetralogy of Fallot (VSD overriding of aorta, pulmonary stenosis, and right ventricular hypertrophy) and patent foramen ovale. The patient had craniofacial and limb abnormalities including mild dysmorphic facial features and polydactyly (one extra digit on both hands and left foot). On genitourinary examination, the patient had solitary kidney and retracted penis. Central nervous system examination revealed microcephaly. Examination of the respiratory tract revealed no evidence of infection.
Discussion
Myocardial sinusoids, the in situ vascular endothelial network, and coronary buds on the aortic sinuses are the basic components needed for the development of coronary vasculature. Abnormal development of any of these components leads to the development of coronary artery anomalies. In the first weeks of fetal development, sinusoids are developed from the inner, trabeculated portion of developing myocardium. Initially, the metabolic exchange between the cardiac cavities and cardiac mesenchyme occurs in the sinusoids. The left proximal part of bulbus cordis gives rise to the left coronary artery bud, while the right proximal part gives rise to the right coronary artery bud [13]. In situ vasculature on the epicardial surface infiltrates the myocardium, with some endocardial contributions as well. After the separation of aorta and pulmonary artery from the bulbus cordis, the in-situ vasculature fuses with the coronary buds, now ostia, to form the coronary circulation that supplies blood to the myocardium [13] (Figure 2).
Due to the embryologic origin of coronary ostia, it is not surprising that coronary ostial abnormalities are more common in conotruncal defects such as tetralogy of Fallot and transposition of the great arteries [13,14]. After simple defects like VSDs and ASDs, conotruncal defects are the most common of the more complex lesions found in trisomy 13 patients [5,6]. Therefore, it would not be unreasonable to expect some trisomy 13 patients to have congenital abnormalities of their coronary ostia. The same would be expected in trisomy 18, which can also see conotruncal defects, though slightly less frequently than in trisomy 13 [5, 6]. In contrast, after VSDs and ASDs, the more common complex lesions found in trisomy 21 are endocardial cushion type defects, or atrioventricular septal defects [15].
In terms of their anatomy, coronary artery anomalies are classified as anomalies of a) origin and course, b) intrinsic coronary artery pathway, and c) termination [16]. Some of these anomalies are benign, having no hemodynamic implications. However, some of the anomalies can cause hemodynamic instability and even sudden cardiac death [16]. Anomalous origin of the coronary arteries from the opposite sinus, anomalous left coronary artery from the pulmonary artery, single coronary artery, intramural coronary artery, and coronary artery fistulas are the high-risk coronary artery anomalies that can cause sudden death [13,17,18]. Coronary ostium anomalies include atresia of coronary ostium and valve-like ridge [13]. Furthermore, ostia can arise from the commissural level and have higher or lower positioning than the normal, as in this case
In our patient, the left coronary ostium showed stenosis and high take off. In addition, it was located above the commissure between the left and posterior aortic cusps. These findings have been associated with sudden cardiac death in neonates [19]. In a retrospective study done by Laux et.al., left coronary ostial stenosis and high take off with acute angle origin was diagnosed at autopsy in 3 infants who died within minutes to days after birth [19]. As the median survival rate for patients with Patau syndrome is 7-10 days, these features could have been masked due to other complications. Our patient was 7 months old, and other causes such as pneumonia were ruled out as the immediate cause of death. We suspect the aberrant coronary artery finding along with the multiple cardiac and non-cardiac comorbidities associated with Trisomy 13 greatly increased the risk of sudden cardiac death for this patient. Patau syndrome is associated with many congenital heart anomalies, but to our knowledge, this is the first report of an association with coronary ostium abnormality. Due to the difficult nature of detecting such lesions clinically, and the low rates of hospital autopsy, it is possible that other Trisomy 13 patients with sudden death have had a similar anatomic lesion. Furthermore, awareness of the possibility of a coronary ostium abnormality, especially in the setting of a conotruncal defect, can help expand the workup of patients who may have unexplained cardiac ischemia and help direct management.
- Williams GM, Brady R (2023) Patau Syndrome. Treasure Island, FL, StatPearls Publishing. PMID: 30855930.
- Goel N, Morris JK, Tucker D, de Walle HEK, Bakker MK, et al. (2019) Trisomy 13 and 18-Prevalence and mortality-A multi-registry population-based analysis. Am J Med Genet A. 179: 2382-92.
- Warburg M (1960) Anophthalmos complicated by mental retardation and cleft palate. Acta Ophthalmol. 38: 394-404.
- Patau K, Smith DW, Therman E, Inhorn SL, Wagner HP (1960) Multiple congenital anomaly caused by an extra autosome. Lancet. 1: 790-3.
- Pont SJ, Robbins JM, Bird TM, Gibson JB, Cleves MA, Tilford JM, Aitken Mev (2006) Congenital malformations among liveborn infants with trisomies 18 and 13. Am J Med Genet A. 150: 1749-56.
- Carey, John C (2021) Trisomy 18 and trisomy 13 syndromes. Cassidy and Allanson's management of genetic syndromes. Hoboken, NJ, John Wiley and Sons, Inc. 937-56.
- Musewe NN, Alexander DJ, Teshima I, Smallhorn JF, Freedom RM (1990) Echocardiographic evaluation of the spectrum of cardiac anomalies associated with trisomy 13 and trisomy 18. Journal of the American College of Cardiology. 15: 673-7.
- Peroos S, Forsythe E, Pugh JH, Arthur-Farraj P, Hodes D (2012) Longevity and Patau syndrome: what determines survival? BMJ Case Rep. bcr0620114381.
- Kato N, Morisaki N, Moriichi A (2024) Trends in the survival of patients with trisomy 13 from 1995 to 2021: A population study in Japan. Am J Med Genet A. 194: e63710.
- Meyer RE, Liu G, Gilboa SM, Ethen MK, Aylsworth AS, Powell CM, et al. (2016) National Birth Defects Prevention Network. Survival of children with trisomy 13 and trisomy 18: A multi-state population-based study. Am J Med Genet A. 170A: 825-37.
- Nelson KE, Rosella LC, Mahant S, Guttmann A (2016) Survival and Surgical Interventions for Children with Trisomy 13 and 18. JAMA. 316: 420-8.
- Rasmussen SA, Wong LY, Yang Q, May KM, Friedman JM (2003) Population-based analyses of mortality in trisomy 13 and trisomy 18. Pediatrics. 111: 777-84.
- Villa AD, Sammut E, Nair A, Rajani R, Bonamini R, Chiribiri A (2016) Coronary artery anomalies overview: The normal and the abnormal. World J Radiol. 8: 537-55.
- Silva A, Baptista MJ, Araújo E (2018) Congenital anomalies of the coronary arteries. Rev Port Cardiol (Engl Ed).
- Peterson JK, Clarke S, Gelb BD, Kasparian NA, Kazazian V, et al. (2024) American Heart Association Pediatric Cardiovascular Nursing Committee of the Council on Cardiovascular and Stroke Nursing, Council on Clinical Cardiology, Council on Genomic and Precision Medicine, Council on Cardiovascular Radiology and Intervention. Trisomy 21 and Congenital Heart Disease: Impact on Health and Functional Outcomes from Birth Through Adolescence: A Scientific Statement From the American Heart Association. J Am Heart Assoc. 13: e036214.
- Pérez-Pomares JM, de la Pompa JL, Franco D, Henderson D, Ho SY, et al. (2016) Congenital coronary artery anomalies: a bridge from embryology to anatomy and pathophysiology--a position statement of the development, anatomy, and pathology ESC Working Group. Cardiovasc Res. 109: 204-16.
- Young PM, Gerber TC, Williamson EE, Julsrud PR, Herfkens RJ (2011) Cardiac imaging: Part 2, normal, variant, and anomalous configurations of the coronary vasculature. AJR Am J Roentgenol. 197: 816-26.
- Angelini P (2002) Clinical articles: coronary artery anomalies—current clinical issues: definitions, classification, incidence, clinical relevance, and treatment guidelines. Texas Heart Institute Journal. 29: 271.
- Laux D, Bessieres B, Houyel L, Bonniere M, Magny JF, Bajolle F, Boudjemline Y, Bonnet D (2013) Early neonatal death and congenital left coronary abnormalities: ostial atresia, stenosis and anomalous aortic origin. Arch Cardiovasc Dis. 106: 202-8.
FIGURE 1
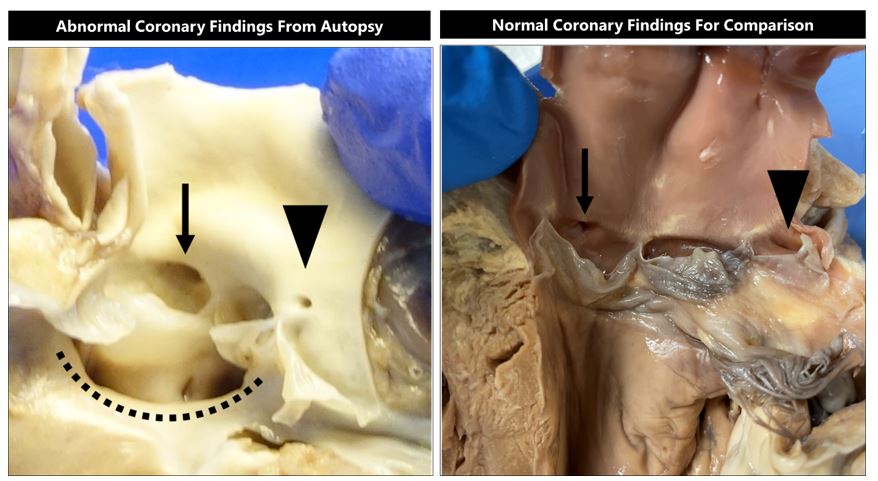
Figure 1: (Left) View of abnormal left ventricular outflow tract showing ventricular septal defect (dotted line), normally positioned right coronary ostium (arrow), and stenotic malpositioned left coronary ostium (arrowhead). (Right) View of normal left ventricular outflow tract showing normally positioned right coronary ostium (arrow) and normally positioned left coronary ostium (arrowhead) for comparison.
FIGURE 2
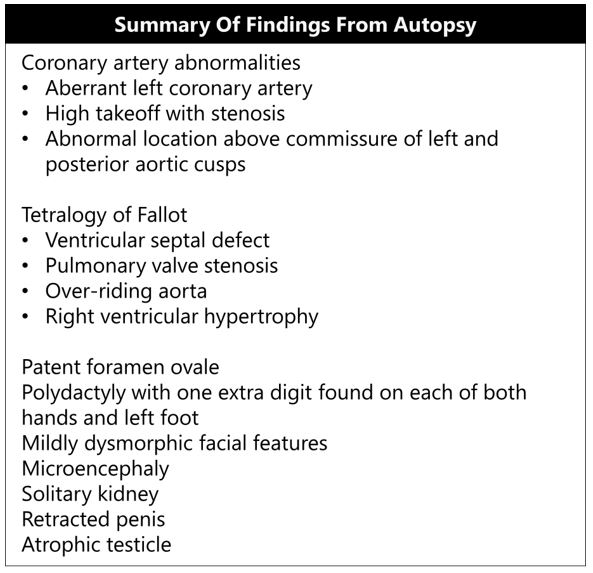
Table 1: Summary of findings from autopsy.
FIGURE 3
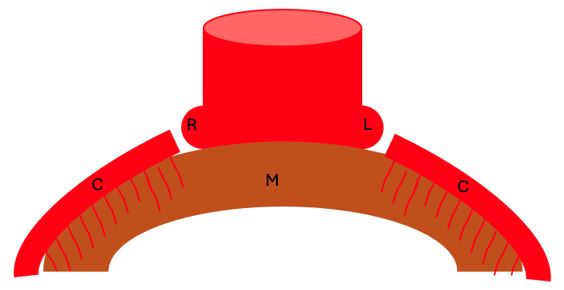
Figure 2: The right (R) and left (L) coronary buds of the aorta merge with in situ coronary vasculature (C) that develops mostly from the epicardial surface to supply the myocardium (M).
Figures at a glance