Unravelling the Genetic Basis and Pathogenic Mechanisms of Primary RCM
Received Date: May 16, 2025 Accepted Date: June 16, 2025 Published Date: June 19, 2025
doi:10.17303/jcvm.2025.11.103
Citation: Rajasekhar Moka, Prabodh Kumar (2025) Unravelling the Genetic Basis and Pathogenic Mechanisms of Primary RCM. J Cardio Vasc Med 11: 1-9
Abstract
Restrictive cardiomyopathy (RCM) is a rare but severe myocardial disorder characterized by impaired ventricular relaxation and reduced diastolic compliance, while systolic function typically remains preserved until advanced stages. Clinically, patients present with progressive dyspnea, fatigue, arrhythmias, and symptoms resembling heart failure with preserved ejection fraction (HFpEF). Secondary RCM commonly arises from systemic or infiltrative diseases such as amyloidosis, sarcoidosis, and haemochromatosis. In contrast, primary RCM is frequently genetic in origin, associated with mutations in genes encoding sarcomeric (e.g., TNNI3, MYH7), cytoskeletal (e.g., DES, FLNC), nuclear envelope (e.g., LMNA), RNA-binding (e.g., RBM20), ion channel, and mitochondrial proteins. These mutations compromise cardiomyocyte integrity, calcium handling, mechanosensation, and bioenergetics, ultimately contributing to diastolic dysfunction and increased arrhythmogenic risk. Diagnosis relies on multimodal imaging, histopathologic evaluation, and genetic testing to distinguish primary from secondary forms. Genetic insights play a pivotal role in guiding family screening, prognostication, and consideration of implantable devices such as ICDs. Ongoing advances in genotype–phenotype correlation and molecular therapeutics are paving the way for precision medicine and improved clinical outcomes in RCM.
Keywords: Restrictive Cardiomyopathy; Diastolic Dysfunction; Sarcomeric Genes; Arrhythmia; Genetic Testing; Genotype–Phenotype Correlation; Heart Failure with Preserved Ejection Fraction (HFpEF); Precision Cardiology
Introduction
Cardiomyopathies encompass a diverse set of myocardial diseases that lead to structural and functional cardiac abnormalities in the absence of ischemic, hypertensive, or valvular pathology. According to major cardiology guidelines by the AHA and the ESC, cardiomyopathies are broadly categorized into five subtypes: dilated, hypertrophic, restrictive, arrhythmogenic right ventricular, and unclassified forms [1,2]. Restrictive cardiomyopathy (RCM), although the least prevalent, is among the most clinically severe types due to progressive diastolic dysfunction and limited treatment modalities [3]. RCM occurs when the heart’s filling is impaired due to increased myocardial stiffness, while systolic function remains preserved until late stages. Key imaging features include non-dilated, non-hypertrophied ventricles with biatrial enlargement. Clinically, RCM presents with features of heart failure with preserved ejection fraction (HFpEF), atrial arrhythmias, thromboembolism, and eventual right-sided heart failure. Etiologically, RCM may be secondary to infiltrative disorders (e.g., amyloidosis, sarcoidosis) or primary (idiopathic or genetic), the latter being the principal focus of this review [4]. Despite its rarity accounting for <5% of cardiomyopathies RCM often follows an aggressive course, especially in children and young adults. Onset ranges from infancy to late adulthood, with symptoms including exertional dyspnea, fatigue, peripheral edema, and arrhythmias. Echocardiographic findings of biatrial enlargement and restrictive filling patterns, along with elevated diastolic pressures, aid diagnosis. Differentiation from constrictive pericarditis remains a crucial clinical step given overlapping phenotype [5].
Genetic studies have shown that primary RCM often involves changes in genes that make proteins important for muscle structure and function, like TNNI3, MYH7, MYBPC3, DES, and FLNC [6]. Accurate identification of these mutations is critical for diagnosis, family screening, risk stratification, and guiding targeted therapy. DNA sequencing and genetic tests are being used more and more, and they are now an important part of diagnosing cardiomyopathies [7].
Clinical Features and Diagnosis
Primary RCM is characterized by impaired diastolic relaxation and elevated ventricular filling pressures, while systolic function remains preserved until later disease stages. Ventricular dimensions remain normal, but the myocardium becomes non-compliant. These changes manifest clinically as progressive exertional dyspnea, orthopnea, fatigue, and systemic and pulmonary venous congestion. Atrial fibrillation is common and contributes to worsening hemodynamics. Pediatric patients and those with genetic forms are at increased risk for syncope and sudden cardiac death [8. 9]. Diagnosis of RCM involves a multimodal approach, incorporating imaging, hemodynamics, and histopathology. Echocardiography is the initial modality of choice, revealing non-dilated ventricles and significant biatrial enlargement. Doppler imaging often demonstrates restrictive filling physiology, characterized by a shortened deceleration time and elevated E/A ratio, indicating increased left ventricular end-diastolic pressure [10].
Cardiac Magnetic Resonance Imaging (CMR) provides further diagnostic precision, particularly for tissue characterization. In primary RCM, CMR typically shows normal-sized ventricles, biatrial enlargement, and minimal or absent late gadolinium enhancement (LGE), unless fibrosis is present. Specific LGE patterns can aid in distinguishing primary RCM from infiltrative etiologies such as amyloidosis or sarcoidosis [11]. Endomyocardial biopsy (EMB) is reserved for diagnostically uncertain cases or when infiltrative disease is suspected. EMB can help exclude secondary causes like amyloid, sarcoidosis, or iron overload [12]. Genetic RCM typically follows an autosomal dominant inheritance pattern, with pathogenic variants in TNNI3, MYH7, DES, FLNC, and LMNA. In contrast, secondary forms arise from conditions like amyloidosis, hemochromatosis, sarcoidosis, or prior chemotherapy. Clinical signs like thicker heart walls, bright heart tissue on ultrasound, overall body effects, or past exposure help doctors categorise the condition [13]. Genetic testing, therefore, plays a pivotal role in resolving diagnostic uncertainty and informing management.
For an accurate diagnosis and appropriate therapy, it is critical to distinguish between primary (genetic) and secondary (induced by other circumstances) restrictive cardiomyopathy. Primary RCM typically follows an autosomal dominant inheritance pattern and is caused by pathogenic variants in genes associated with sarcomeric contractility (e.g., TNNI3, MYH7), cytoskeletal stability (e.g., DES, FLNC), and nuclear envelope integrity (e.g., LMNA) [4,6]. In contrast, secondary RCM happens when the heart muscle is affected or damaged by other health issues like cardiac amyloidosis, hemochromatosis, sarcoidosis, or fibrosis after radiation treatment. Clinical indicators of secondary forms include increased ventricular wall thickness, echogenic myocardium, systemic features like neuropathy or hepatomegaly, and a history of chemotherapy or radiotherapy exposure [11,13]. In phenotypically ambiguous cases, comprehensive genetic testing not only enhances diagnostic confidence but also aids prognostication and facilitates cascade screening among relatives at risk [7].
Genetic Etiology of Primary RCM
Restrictive cardiomyopathy (RCM) is increasingly recognized as a genetically heterogeneous disorder. Many primary cases arise from pathogenic variants in genes affecting sarcomeric proteins, cytoskeletal structure, the nuclear envelope, mitochondria, and ion-handling mechanisms. These mutations compromise myocardial relaxation, promote fibrosis, and increase arrhythmogenic risk, often clustering in families [14,15]. Key genes and their functional classifications are summarized in Table 1, and specific genotype–phenotype correlations are detailed in Table 2.
Sarcomeric Genes
Sarcomeric gene mutations are the most common cause of primary RCM. These genes encode contractile proteins essential for myocardial contraction and relaxation. Variants in TNNI3 and TNNT2, encoding troponin I and T respectively, increase calcium sensitivity, impair relaxation, and contribute to ventricular stiffening [16]. While MYH7 and MYBPC3 are typically associated with hypertrophic or dilated cardiomyopathies, specific mutations such as MYH7 p.Arg145Gly have been identified in familial RCM with early-onset heart failure and sudden cardiac death (SCD) [17,18]. These mutations are usually inherited in an autosomal dominant manner with variable expressivity. Notably, TNNI3 p.Arg145Gly is a recurrent mutation hotspot associated with adverse outcomes [19].
Cytoskeletal and Z-Disk Proteins
Cytoskeletal and Z-disk proteins are vital for maintaining myocardial structure and transmitting mechanical forces. Mutations in DES, encoding desmin, lead to disorganized intermediate filaments and desmin-positive aggregates, manifesting as RCM with skeletal myopathy and conduction defects [18]. Likewise, truncating mutations in FLNC (filamin C) impair actin binding, contribute to myocardial fibrosis, and increase susceptibility to ventricular arrhythmias and SCD [20]. ACTN2, encoding α-actinin-2, is typically implicated in hypertrophic or dilated cardiomyopathies, but certain variants present with restrictive features, further expanding the phenotypic spectrum of sarcomere-related disorders [21].
Nuclear Envelope and RNA-Binding Proteins
Mutations in nuclear envelope proteins, particularly LMNA, disrupt nuclear architecture and mechanosensitive gene regulation. LMNA-related cardiomyopathies frequently manifest with conduction disease, arrhythmias, and myocardial fibrosis, including RCM phenotypes [22]. In Indian families, LMNA mutations have been implicated in early-onset cardiomyopathies with high penetrance and familial SCD [14]. RBM20, an RNA-binding protein regulating titin (TTN) and RYR2 splicing, is another gene of interest. Mutations lead to mis-splicing of sarcomeric proteins, impaired relaxation, and fibrotic remodeling. Although more commonly linked to dilated cardiomyopathy, RBM20 variants have been identified in RCM, reflecting overlapping pathophysiological mechanisms [23,24].
Mitochondrial and Ion-Handling Genes
Mitochondrial bioenergetics and calcium homeostasis are critical for myocardial relaxation. Mutations in TAZ, which encodes tafazzin and is associated with Barth syndrome, result in deficient cardiolipin biosynthesis. This disrupts mitochondrial structure and function, leading to energy failure and early-onset cardiomyopathy with restrictive features, particularly in pediatric cases [25]. Similarly, mutations in PLN (phospholamban), a regulator of sarcoplasmic reticulum Ca²⁺ ATPase (SERCA2a), impair calcium reuptake, compromising diastolic relaxation. PLN mutations have been reported in both dilated and restrictive cardiomyopathy and are associated with arrhythmogenic phenotypes [26].
Additionally, mutations in SCN5A and RYR2, both ion-handling genes, can contribute to RCM through arrhythmogenesis and diastolic dysfunction. SCN5A encodes a cardiac sodium channel, while RYR2 encodes the ryanodine receptor involved in calcium release from the sarcoplasmic reticulum. Their pathogenic variants are increasingly recognized as contributors to restrictive physiology via altered excitation-contraction coupling [24].
Non-Coding RNAs and Epigenetic Mechanisms
Beyond protein-coding mutations, non-coding RNAs (ncRNAs)—including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs)—play key regulatory roles in the molecular pathogenesis of restrictive cardiomyopathy (RCM). miRNAs act as post-transcriptional repressors of mRNA targets involved in sarcomeric organization, calcium handling, and myocardial fibrosis. Notably, miR-133 suppresses collagen synthesis by targeting TGF-β pathway components, and its downregulation contributes to fibrotic remodeling, while miR-208 promotes hypertrophic and fibrotic gene programs by modulating MYH7 expression [27,28]. Additionally, studies on human heart disease have found that the levels of certain miRNAs and mRNAs in the heart can affect each other, and some of these changes can be reversed when the heart is relieved from stress, showing their role in how the disease progresses [29]. LncRNAs such as MHRT protect the heart from pathological stress by interfering with BRG1-mediated chromatin remodeling, while other lncRNAs like FENDRR and ANRIL are involved in transcriptional regulation and cardiac development [30].
Epigenetic modifications including DNA methylation and histone modifications, mediate gene-environment interactions in idiopathic RCM. Aberrant promoter methylation of cardiac genes can lead to impaired calcium cycling and altered sarcomeric protein expression. External triggers such as oxidative stress and metabolic dysregulation may induce or exacerbate these epigenetic shifts, potentially accounting for phenotypic variability and reduced penetrance in familial cases [31,32].
Genotype–Phenotype Correlations and Clinical Implications
Understanding genotype–phenotype correlations in restrictive cardiomyopathy (RCM) is essential for improving diagnosis, prognostication, family counselling, and therapeutic decision-making. Genetic changes in RCM show a wide range of symptoms, even in people with the same mutation, which emphasises how symptoms can vary and not everyone with a mutation will have the same issues [33,34].
For example, changes in the TNNI3 gene, which makes cardiac troponin I, often lead to early heart problems, a stiff heart, and a higher chance of SCD [6,35]. These patients need careful medical supervision, and getting an implantable cardioverter-defibrillator (ICD) might be necessary for those with a family history of severe heart failure or brief episodes of fast heartbeats. Notably, the same TNNI3 mutation can cause asymptomatic disease in one individual and SCD in another [33]. Mutations in LMNA and FLNC can result in significant cardiac arrhythmias, frequently disrupting the conduction of electrical signals inside the heart and resulting in tachycardia, necessitating early consideration of assistive devices by physicians [34,36]. Patients with DES mutations may show problems in both their skeletal and heart muscles, which means they need care from both heart doctors and muscle specialists [37].
Some variants such as in ACTN2 and RBM20 may result in later-onset or subtler disease but require cascade screening and early monitoring of asymptomatic relatives due to autosomal dominant inheritance [38,39]. Furthermore, genotype can assist in differentiating genetic RCM from phenocopies. For example, cardiac amyloidosis and hemochromatosis may mimic restrictive physiology but require fundamentally different therapies. Advanced imaging techniques, like cardiac MRI with T1 mapping, along with genetic testing, make it easier to diagnose conditions accurately and can help find patients who might benefit from new treatments targeted at specific mutations [11,13].
Targeted Therapies and Genotype-Guided Management in RCM
As the genetic basis of restrictive cardiomyopathy (RCM) becomes clearer, new avenues for targeted and precision therapies are emerging. While supportive and device-based management remain the mainstay, genotype-driven interventions are increasingly being explored. Myosin inhibitors such as mavacamten and aficamten, initially developed for hypertrophic cardiomyopathy, are now being investigated in early-phase trials for their potential to reduce sarcomeric hypercontractility and improve diastolic function in select RCM genotypes, particularly those involving MYH7 mutations [40]. Antisense oligonucleotide (ASO) therapies are a promising molecular tool aimed at modifying gene expression. For example, researchers are working on ASOs that focus on LMNA transcripts to lower harmful protein buildup and reduce heart rhythm problems in LMNA-related cardiomyopathy [41]. Gene therapy methods such as exon skipping are being studied for TTN mutations that cause truncation, which helps to fix the reading frame and partially restore titin function [42]. Though these therapies are not yet clinically approved for RCM, they represent a shift toward precision medicine in inherited cardiomyopathies.
Importantly, management strategies differ by genotype. For example, people with LMNA mutations are at a high risk for dangerous heart rhythm problems and usually need to have an implantable cardioverter-defibrillator (ICD) put in early, even if their left ventricle isn't severely damaged [43]. On the other hand, patients with TNNI3 or FLNC mutations might need regular check-ups for heart issues and irregular heartbeats, which will help decide on medication and device treatments [44]. As genetic therapies continue to evolve, incorporating genotype-specific risk stratification and targeted treatment will be key to optimizing outcomes in RCM [45].
Future Perspectives and Conclusion
Despite its rarity, restrictive cardiomyopathy (RCM) presents substantial clinical obstacles as a result of its genetic heterogeneity and progressive nature. Advancements in genomic technologies have enhanced the accuracy of early diagnosis, risk stratification, and family screening. Future endeavours should prioritise the identification of novel therapeutic targets, epigenetic modifiers, and genotype-specific mechanisms. Gene editing, antisense therapies, and integrated multi-omic profiling are emerging approaches that demonstrate potential for personalised treatment. Finally, it will be imperative to implement a collaborative approach that integrates genetic, clinical, and imaging data in order to transform scientific discoveries into improved outcomes for patients with RCM.
- Maron BJ, Towbin JA, Thiene G, et al. (2006) Contemporary definitions and classification of the cardiomyopathies. Circulation. 113: 1807-16.
- Elliott P, Andersson B, Arbustini E, et al. (2008) Classification of the cardiomyopathies: a position statement from the ESC Working Group. Eur Heart J. 29: 270-6.
- Panicker GK, Karnad DR, Natekar M, et al. (2015) Cardiomyopathies in India: Clinical features and spectrum. Indian Heart J. 67: 219-24.
- Kumar S, Shetty R, Ghosh S, et al. (2019) Clinical and genetic profile of restrictive cardiomyopathy in Indian patients. Indian Heart J. 71: 121-7.
- Saxena A, Deshpande S, Pant P, et al. (2018) Genetic mutations in hypertrophic cardiomyopathy: Indian perspective. J Genet. 97: 1157-64.
- Kubo T, Gimeno JR, Bahl A, et al. (2002) Prevalence and prognosis of troponin T mutations in familial HCM. J Am Coll Cardiol. 39: 286-94.
- Gupta A, Mohan JC, Seth S, et al. (2015) Clinical profile and outcome of idiopathic restrictive cardiomyopathy: an Indian experience. Indian Heart J. 67: 123-8.
- Ammirati E, Contri R, Cipriani M, et al. (2017) Clinical presentation and outcome in a contemporary cohort of patients with RCM. Heart. 103: 766-72.
- Brion M, Rubio M, Alonso B, et al. (2010) Molecular basis of restrictive cardiomyopathy. Rev Esp Cardiol. 63: 827-31.
- Nagueh SF, Appleton CP, Gillebert TC, et al. (2009) Recommendations for the evaluation of LV diastolic function by echocardiography. Eur J Echocardiogr. 10: 165-93.
- Miller CA, Naish JH, Bishop P, et al. (2013) Validation of CMR techniques for assessing myocardial extracellular volume. Circ Cardiovasc Imaging. 6: 373-83.
- Choudhury SR, Kumar S, Sahu A, et al. (2016) Role of biopsy in cardiomyopathies: Indian experience. Indian J Pathol Microbiol. 59: 354-9.
- Phelan D, Collier P, Thavendiranathan P, et al. (2014) The role of cardiac imaging in the evaluation of heart failure. JACC Cardiovasc Imaging. 7: 239-54.
- Rao VS, Chandrasekaran S, Arumugam K, et al. (2017) Genetics of cardiomyopathy: Insights from Indian families. Indian J Hum Genet. 23: 230-7.
- Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P. (2016) Increasing role of titin mutations in neuromuscular disorders. J Neuromuscul Dis. 3: 293-308.
- Mogensen J, Murphy RT, Shaw A, et al. (2004) Severe disease expression of cardiac troponin C and I mutations in patients with idiopathic restrictive cardiomyopathy. J Clin Invest. 114: 396-403.
- Frustaci A, Chimenti C, Ricci R, et al. (1996) Histological and molecular diagnosis of idiopathic restrictive cardiomyopathy. Am J Cardiol. 77: 1074-80.
- Dellefave-Castillo LM, Gajewski D, Ferrero-Miliani L, et al. (2011) Cardiomyopathy in a family with a novel TNNI3 mutation. Am J Med Genet A. 155A: 2247-52.
- Pugh TJ, Kelly MA, Gowrisankar S, et al. (2014) The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 16: 601-8.
- Begay RL, Tharp CA, Martin A, et al. (2018) FLNC gene splice mutations cause arrhythmogenic cardiomyopathy. Circ Genom Precis Med. 11: e001663.
- Chiu C, Bagnall RD, Ingles J, et al. (2010) Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: A genome-wide analysis. J Am Coll Cardiol. 55: 1127-35.
- van Berlo JH, de Voogt WG, van der Kooi AJ, et al. (2005) Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations. J Mol Med. 83: 79-83.
- van Spaendonck-Zwarts KY, van Rijsingen IA, van den Berg MP, et al. (2013) Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: Overview of 10 years’ experience. Eur J Heart Fail. 15: 628-36.
- Refaat MM, Lubitz SA, Makino S, et al. (2012) Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm. 9: 390-6.
- Xu Y, Malhotra A, Ren M, et al. (2006) Ablation of tafazzin gene expression in mice leads to mitochondrial abnormalities and early lethality. Mol Cell Biol. 26: 4143-52.
- van der Zwaag PA, van Rijsingen IA, de Ruiter R, et al. (2012) Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy. Eur J Heart Fail. 14: 1199-207.
- Care A, Catalucci D, Felicetti F, et al. (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med. 13: 613-8.
- van Rooij E, Sutherland LB, Qi X, et al. (2007) Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 316: 575-9.
- Matkovich SJ, Van Booven DJ, Youker KA, et al. (2009) Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy. Circulation. 119: 1263-71.
- Han P, Li W, Lin CH, et al. (2014) A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 514: 102-6.
- Greco CM, Condorelli G. (2015) Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol. 12: 488-97.
- Movassagh M, Choy MK, Knowles DA, et al. (2011) Distinct epigenomic features in end-stage failing human hearts. Circulation. 124: 2411-22.
- Kostareva A, Gudkova A, Sjoberg G, et al. (2009) De novo mutation in the cardiac troponin I gene in a newborn with restrictive cardiomyopathy. Cardiovasc Pathol. 18: 362-4.
- Taylor MR, Fain PR, Sinagra G, et al. (2007) Natural history of dilated cardiomyopathy due to troponin T and I mutations. J Am Coll Cardiol. 49: 2606-13.
- Hasselberg NE, Haland TF, Saberniak J, et al. (2018) Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for implantable cardioverter defibrillator. Eur Heart J. 39: 853-60.
- van Rijsingen IA, Arbustini E, Elliott PM, et al. (2012) Risk factors for malignant ventricular arrhythmias in Lamin A/C mutation carriers. J Am Coll Cardiol. 59: 493-500.
- Clemen CS, Herrmann H, Strelkov SV, Schröder R. (2013) Desminopathies: pathology and mechanisms. Acta Neuropathol. 125: 47-75.
- Bagnall RD, Molloy LK, Kalman JM, Semsarian C. (2014) Genetic basis of familial atrial fibrillation. J Am Coll Cardiol. 64: 2817-26.
- Maatz H, Jens M, Liss M, et al. (2014) RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest. 124: 3419-30.
- Olivotto I, Oreziak A, Barriales-Villa R, et al. (2020) Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomized, double-blind, placebo-controlled, phase 3 trial. Lancet. 396: 759-69.
- Bonne G, Muchir A. (2022) LMNA-targeted antisense therapy for laminopathies: preclinical promise. J Clin Invest. 132: e160102.
- Swinnen B, Vlaminck L, de Wit E, et al. (2021) Exon-skipping therapy in titin truncation-related dilated cardiomyopathy: a proof-of-concept study. Circulation. 143: 1731-43.
- Hershberger RE, Givertz MM, Ho CY, et al. (2018) Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail. 24: 281-302.
- Walsh R, Thomson KL, Ware JS, et al. (2017) Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 19: 192–203.
- McNally EM, Mestroni L. (2017) Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 121: 731-48.
FIGURE 1
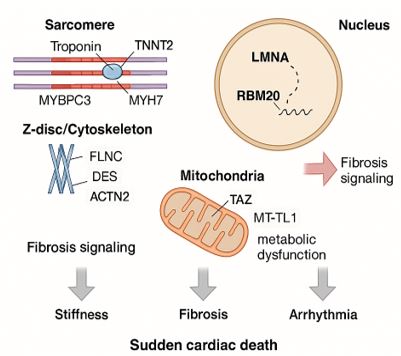
Figure 1: Molecular pathways and cellular structures are affected in RCM
This schematic illustrates the key molecular components and cellular structures implicated in the pathogenesis of restrictive cardiomyopathy. The sarcomere is shown with contractile proteins including MYH7, MYBPC3, and troponins (TNNI3, TNNT2), reflecting impaired myocardial contraction. The Z-disc/cytoskeleton is represented by FLNC, DES, and ACTN2, proteins critical for structural integrity and mechanotransduction. The nucleus features LMNA and RBM20, linked to nuclear envelope stability and transcriptional regulation. Mitochondrial dysfunction is illustrated by TAZ and MT-TL1, associated with impaired energy metabolism. The fibrotic response is highlighted through the TGF-β signaling pathway, a central mediator of myocardial fibrosis. Arrows depict downstream pathological consequences including increased myocardial stiffness, fibrotic remodeling, arrhythmias, and the risk of sudden cardiac death.
Tables at a glance
Figures at a glance