Comprehensive Identification of RNA Transcripts and Construction of RNA Networks in Coronary Artery Calcification
Received Date: June 06, 2023 Accepted Date: July 06, 2023 Published Date: July 10, 2023
doi: 10.17303/jcvm.2023.9.101
Citation: Hao Hu, Zhen Wang, Yucong Wang, Liang Chen, Likun Ma (2023) Comprehensive Identification of RNA Transcripts and Construction of RNA Networks in Coronary Artery Calcification. J Cardio Vasc Med 9: 1-22
Abstract
Coronary artery calcification (CAC) frequently occurs in patients with coronary heart disease, strongly predicting risk for future cardiovascular events. However, it could be detected until it increases in quantity. Roles of aberrant expression of RNAs in CAC have been proposed, yet limited studies have systematically investigated the crosstalk among various RNA transcripts. Here, we have comprehensively identified and analyzed the differentially expressed mRNAs, lncRNAs, miRNAs and circRNAs in peripheral blood of CAC patients with high-throughput RNA sequencing. Gene Set Enrichment Analysis (GSEA) showed that these dysregulated RNAs correlate with several critical biological processes such as RNA modification,and RNA metabolism. RT-qPCR with 30 pairs of clinical CAC samples were used for the validation of some differentially expressed RNAs. We have also constructed the co-expression network between lncRNA and mRNA, and for the first time established the circRNA-miRNA-mRNA in CAC. Predicted functional mechanism of the circRNAs as miRNA sponges were further validated and discussed with AGO2 RNA immunoprecipitation (RIP). This study provides valuable prospective insights to understand the RNA involvement and regulation in CAC for future in-depth investigations.
Keywords: Coronary Artery Calcification; MRNA; LncRNA; CircRNA; RNA Network
Introduction
Coronary artery calcification (CAC) predicts high morbidity and mortality risk worldwide, which is characterized by pathological deposition of minerals in the artery wall and is currently considered to be related to atherosclerosis [1-6]. The degree of coronary artery calcification correctly assigns patients into low-risk and high-risk categories.Patients with high level of coronary calcification is commonly observed in almost all patients with coronary artery disease (CAD), which is a significant feature for coronary artery disease events [6,7]. The biogenesis of CAC occurs at the very early stage of atherosclerosis, but it can be hardly detected before it increases in quantity and is observed with professional imaging systems. The CAC scoring (CACS) has been widely used as a measure that could help with risk assessment and cardiac event prediction, with advantages of directly visualizing and precisely detecting the plaques using computed tomography (CT), making it as a surrogate marker for atherosclerotic plaque burden [8,9].
Unlike conventional risk factors that could only provide a statistical probability for patients developing CAD, CAC scoring can also be assessed using Agatston calcium scoring,enabling a direct evaluation of each patient on an individual basis [10]. Compared with the conventional coronary angiography (CCA), which is invasive, expensive, and inconvenient for patients, CAC scoring is currently more used in the routine clinical practice as a noninvasive technique.CAC scoring of certain degree helps reclassify the risk of many patients and estimate future cardiovascular events,and that of greater than 400 is associated with worse clinical outcomes. However, these techniques only provide reliable prediction at early stages of existing CAD with prognosis implications, and requires professional clinical input. The genetic basis of CAC for both prognostic and diagnostic application still need to be elucidated. Specific and effective biomolecules for CAC early clinical prevention and treatments is still lacking.
With the advent of the next-generation sequencing,dysregulated RNA has recently revealed its significance in multiple human diseases such as various cancers, neurodegenerative diseases, cardiovascular diseases, etc.[11-13]. It is widely acknowledged that less than 2% of human genome is gene-coding, while the vast majority of human genome is non-coding. Varying mRNAs, miRNAs, lncRNAs and circRNAs are reported to be involved in biogenesis and progression of cardiovascular calcification, with some of them have long regarded as potential clinical predictors in CAC treatments. For example, Genome-wide association studies (GWASs) further suggest that CAC is highly heritable, with genetic cause of CAC is increasingly being recognized [14]. Genes encoding for ionic channel subunits,inflammation, and connexin that involve in CAC have been constantly discovered over the last few decades [15-17]. TREML4, screened from peripheral blood with RNA-sequencing,was upregulated in CAC, and it colocalized with areas of microcalcification within coronary plaques [18]. A series of CAC-related genes have also been identified in gene expression microarrays and cell culture replication in a cell-based investigations [19].
MiRNAs are critical regulators involved in multiple physiological and pathological processes, including development, organogenesis, apoptosis, cell proliferation and differentiation [20]. Dysregulation of miRNA often leads to impaired cellular function and disease progression, including CAC. MiRNA-32a has been reported to be upregulated in CAC and it functions by activating phosphoinositide 3-kinase (PI3K) signaling and increasing RUNX2 expression and phosphorylation in mouse vascular smooth muscle cell (VSMC) [21]. Also, a series of circulating miRNAs were found to be closely related to the level of CAC in symptomatic patients [22].
Long noncoding RNAs (lncRNAs) play crucial roles in numerous biological processes, including epigenetic regulation, imprinting, cell-cycle, cellular differentiation,splicing, nuclear/cytoplasmic trafficking, and transcription/-translation [23]. Recent studies have also suggested critical roles of lncRNAs in modulating the initiation and progression of cardiovascular diseases, including myocardial infarction,heart failure, and atherosclerosis [24-27]. Lrrc75a-as1 could attenuate rat vascular smooth muscle cells calcification via decreasing the expression of osteoblast-related factors [28]. Long noncoding RNA LIPCAR was found downregulated early after myocardial infarction but upregulated during later stages which could potentially predict future death in patients with heart failure [26]. LncRNAs were also identified as proper and specific biomarkers for coronary artery diseases [29].
CircRNAs are a special class of noncoding RNAs with a covalently closed loop through 5’ to 3’ ends generally via backsplicing in animals, which are involved in various diseases including coronary artery diseases [30-32]. Recent advances indicate that circRNAs might function with multiple mechanisms, such as miRNA sponge, RNA-binding protein (RBP) scaffold, transcriptional regulation [31]. Dysregulation of circRNAs were observed in the progression of coronary artery diseases. For example, hsa_circ_0004104 is upregulated in coronary artery disease patients, and it might contribute to [33]. Interestingly, hsa_circ_0004104 is downregulated in the persistency of atrial fibrillation and could be a potential regulator and biomarker [34]. A heart-related circRNA (HRCR) functions as miR-223 sponge to compromise its activity and hence increase ARC expression, a target for miR-223,indicating that it could be used as an attractive therapeutic target for cardiac hypertrophy and heart failure [35]. Circ--Foxo3 interacts with transcriptional factors such as ID-1,E2F1, FAK, and HIF1α and suppresses their anti-senescent and anti-stress roles, leading to increased cellular senescence [36].
The aim of this study is to explore the differentially expressed mRNAs, lncRNAs, miRNAs and circRNAs during the process of CAC, and tentatively reveal the interactions and potential functional mechanisms of the RNAs.We hypothesized that certain dysregulated RNAs could exert their functions either individually or cooperatively, and some of them could be exploited as potential biomarkers.Here, we have comprehensively identified differentially expressed mRNAs, miRNAs, lncRNAs, and circRNAs in peripheral blood of CAC patients through high-throughput RNA sequencing. GSEA analysis showed that these dysregulated RNAs correlate with several critical biological processes= such as RNA modification, RNA metabolism, and RNA processing. To demonstrate the potential interactions between lncRNAs and their interacting mRNAs, we have constructed the lncRNA-mRNA co-expression network in CAC. For the validated circRNAs, we have analyzed the features of the circRNAs and predicted their potential functional mechanisms in CAC. We further for the first time established the circRNA-miRNA-mRNA network in CAC, and validated with AGO2 RIP, which provides valuable prognostic or predictive resource for future CAC research and clinical treatments.
Methods
Clinical Samples
All fresh peripheral blood samples of four patients with CAC and four non-CAC patients from The First Affiliated Hospital of University of Science and Technology of China, which was approved by the Human Research Ethics Committee of University of Science and Technology of China (2019KY ER No.165). Written informed consent was obtained from each patient for this study. All patients enrolled have no chronic diseases such as diabetes, hypertension or renal inadequacy. All methods were carried out in accordance with relevant guidelines and regulations (Declaration of Helsinki).
CAC Scoring
CAC scoring was performed by multislice computed tomography (CT) (Revolution 256 Slice CT, General Electric Healthcare; Software: ADw 4.2) using prospective electrocardiographic gating and the Agatston scoring method [37]. A factor of 1 (+) for 130–199 H, a factor of 2 (++) for 200–299 H, a factor of 3 (+++) for 300-399 H, and a factor of 4 (++++) for densities greater than 400 H. The total calcium score was calculated as the sum of the individual lesion scores in all coronary arteries. All patients in normal control group were diagnosed with total calcium score less than 130 H. All patients in CAC group were diagnosed with total calcium score more than 400 H. Coronary angiography was performed to further confirm the CAC severity, and thus calcification and normal groups were classified. Information of patients for RNA-seq was listed in Table S1. Information of patients for RNA qPCR validation was listed in Table S2.
Cell Culture
HASMC cells originated from the ATCC and were cultured with DMEM (Gibco). All cells were cultured under standard conditions including 10% FBS and 1% penicillin/streptomycin at 37°C under 5% CO2. Cells were tested for mycoplasma by DAPI staining, to ensure the absence of contamination.
Total RNA Extraction
The peripheral blood samples and TRizol reagent (Invitrogen, USA) are evenly mixed in a ratio of 1:3, total of 12 ml. Total RNA was extracted by using TRizol reagent according to the manufacturer’s instructions.
Total RNAs from four pairs of patients with CAC and without CAC blood samples were extracted for high throughput sequencing. Whole transcriptome libraries were constructed by the TruSeq Ribo Profile Library Prep Kit (Illumina, United States), according to the manufacturer’s instructions. In brief, 10 mg total RNA was depleted rRNA with an Illumina Ribo-Zero Gold kit and purified for end repair and 50-adaptor ligation. Then, reverse transcription was performed with random primers containing 30 adaptor sequences and randomized hexamers. Finally, the cDNAs were purified and amplified with thermo cycler. The PCR products of 300–500 bp were purified, quantified and stored at -80 before sequencing. The libraries were subjected to 151 nt paired-end sequencing with an Illumina Nextseq 500 system (Novogene, China).
The high-throughput sequencing tools, Hisat2 [38] and featureCounts [39], were used to map clean reads to Homo sapiens reference genome (hg19) and calculate the gene expression level which was normalized to z-score. To determine the differentially expressed mRNA and lncRNA, the “DEseq2” [40] package in R software was used with the corresponding cutoff (q < 0.05, |log2(fold change)| > 1 for mRNA and lncRNA). To predict circular RNA (circRNA) expression, we identified the possible candidates with find_- circ [41] with the junction reads were normalized to z-score. Standard of P < 0.05, and | log2(fold change)| > 1 was used to identify differentially expressed circRNAs. Cancer- Specific CircRNA database (CSCD, https://gb.whu. edu.cn/CSCD/) was used for the prediction of microRNA response element (MRE) and RNA binding protein (RBP) among differentially expressed circRNAs.
Small RNA Data Analysis
For small RNA (sRNA) sequencing, eight sRNA libraries were generated with TruSeq small RNA (Illumina,United States) according to the manufacturer’s instructions.Then the prepared libraries were sequenced with an Illumina Nextseq 500 system (Novogene, China). After filtering out the reads shorter than 15 nt, the remaining reads were mapped to the human genome (hg19) and the miRNA database in miRBase with bowtie (-v 1) [42]. The differentially expressed miRNAs were determined by DEseq2 [37] with the cutoff of P < 0.05, TPM1, |log2(fold change)|≧1.
Construction of Co-expression and ceRNA Network
For the co-expression network of significantly dysregulated lncRNAs and mRNAs, Pearson’s correlations were used to calculate the co-expression analysis according to the expression level. A criterion of the coefficient parameter R-squared more than 0.99 was used for the remaining RNAs to further construct the network. For the competing endogenous RNAs (ceRNA) network of significantly dysregulated circRNAs and mRNAs, the miRNA/mRNA and miRNA/ circRNA interaction were predicted with TargetScan-Human7.2 [43]. The above networks were both performed with Cytoscape [44].
Gene Set Enrichment Analysis (GSEA)
The GSEA analysis was performed to annotate the function of protein-coding genes (PCGs) and lncRNAs in blood of CAC patients. These PCGs and lncRNAs were preranked and the GSEA analysis was implemented in GSEA software (version 4.1.0, https://www.gsea-msigdb.org) [45,46].
RNA Immunoprecipitation
AGO2 antibody (SAB4200085, Sigma) was incubated with Dynabeads Protein G (10003D, Invitrogen) according to the manufacturer’s recommendations [47]. Cells were harvested 48 hours post transfection and washed twice with PBS and fixed with 1% formaldehyde. RIPA buffer (50 mmol/L Tris-HCl, pH8.0, 150 mmol/L NaCl, 5 mmol/L EDTA, 1% NP-40, 0.1% SDS) was used to lysis the cells followed by sonication for 10 min and centrifugation at 13,000×g for 15 min at 4°C. Cell lysate was preserved and then incubated with antibody coupled beads for the immunoprecipitation.After 4 hours incubation, beads were washed three times with RIPA buffer and 100μl of the samples were preserved for Western blot analysis. The rest was subjected to TRIzol RNA extraction followed by standard reverse transcription (RT) and quantitative realtime PCR (qPCR). Oligos used are shown in Table S3.
Reverse Transcription and Real-Time Quantitative PCR
cDNA was synthesized using GoScript Reverse Transcription System (Promega, USA) according to the manufacturer’s protocol. Quantitative real-time PCR was performed with GoTaq SYBR Green qPCR Master Mix (Promega, USA) on a PikoReal 96 real-time PCR system followed by 40 amplification cycles (Thermo Fisher Scientific,USA) according to standard procedures. Actually, all amplification curves already reached stationary stage before 35 amplification cycles, and the Ct value were obtained at the exponential stage. Relative RNA level was normalized to GAPDH mRNA. All primers are shown in Table S3.
Availability of Data and Materials
The datasets generated and analyzed during the current study are available in the in the GEO (Gene Expression Omnibus). The accession number is GEO: GSE194304
Statistical Analysis
In order to determine whether the data follows normal distribution, the nortest (v 1.0.4) R package was used in all experiments. Student’s t-tests were used to calculate P-values by t.test function in R software, as indicated in the figure legends. The values reported in the graphs represent average of actual number of independent experiments,with error bars showing SD. To calculate correlation coefficients between all groups, the cor function in R software was used. The cluster analysis in heatmaps was conducted by cluster (v 2.1.2) R package and the heatmaps were generated by pheatmap R package (v 1.0.12). The bar plots and scatter plots were generated by ggplot2 (v 3.3.3). After analysis of variance with F-tests with R software, the statistical significance and P-values were evaluated with Student’s t-tests.
Results
This study aimed to use bioinformatics analysis to investigate the differentially expressed mRNAs, lncRNAs,miRNAs and circRNAs in CAC to unveil the potential dynamic changes of both coding and noncoding RNAs. GSEA analysis revealed that multiple RNA-related pathways might be involved in the progression of CAC. Further, qRT-PCR was implemented to validate the RNAs in 30 pairs of CAC samples and the validations are in accordance of the sequencing results with high confidence. We have also established mRNA-lncRNA co-expression network and circRNA-miRNA-mRNA network to unveil the potential interacting mechanisms of these RNAs in CAC, which may help elucidate the functions and functional mechanisms of these RNA candidates in the future, and provide potential targets for both diagnosis and treatments.
Identification of Differentially Expressed Rnas in CAC
To identify CAC-related RNAs, we used fresh peripheral blood samples from four CAC patients for RNA sequencing.Fresh peripheral blood from four patients without calcification in coronary artery were used as controls. Three types of RNA libraries were constructed to further investigate the differentially expressed RNAs and their respective networks in CAC (Figure 1a). More specifically, lncRNA library was constructed for long noncoding RNAs and mRNAs, small RNA library was constructed for microRNAs (miRNAs) (Figure 1a). RNase R digestion is the gold standard for the verification and enrichment of circRNAs as it degrades all linear RNAs with short 3’ tails but does not degrade lariat or circular forms. CircRNA library was hence constructed followed by RNase R treatment (Figure 1a). The differentially expressed mRNA, lncRNAs, circRNAs,and miRNAs in CAC were analyzed. Pearson’s correlation coefficient analysis was used to evaluate the relationship between CAC groups and the control groups, which revealed that CAC groups were positively correlated with each other, and control groups also showed strong positive correlation (Figure 1b).
Analyses of the Differentially Expressed mRNAs and lncRNAs
We first set out to more specifically investigate the differentially expressed mRNAs and lncRNAs with filtration criteria (fold changes ≥2.0 and p-values ≤0.05). Hierarchical clustering heatmap was performed to evaluate these RNAs, in which CAC and control samples were respectively classified into different branches (Figure 2a,b). We totally identified 238 mRNAs and 155 lncRNAs that were differentially expressed. Among them, 91 mRNAs (38.24%) and 64 lncRNAs (41.29%) were upregulated. 147 mRNAs (61.76%) and 91 lncRNAs (58.71%) were downregulated. It seems evident that the four CAC samples differed more from normal samples than among themselves, presumably due to the individual heterogeneity. Accumulative evidence suggest that functions of lncRNAs mainly exert their functions through regulation of the co-expressed protein-coding gene expression with various mechanisms. Hence lncRNA-mRNA co-expression network was constructed to reveal the co-regulatory relationship network of CAC based on their potential multi-reciprocal interactions, which provided the potential functional interactions of lncRNA-mRNA for future studies (Figure 2c).
GSEA Analysis of the Differentially Expressed mRNAs
To investigate the potential pathways and biological processes that these dysregulated RNAs involve in, we performed GSEA analysis with either upregulated or downregulated differentially expressed RNAs (Figure 3a). The results demonstrated that they were closely related to certain critical pathways that might impact the development of CAC such as tRNA metabolic process, ribosomal large subunit biogenesis, innate immune response in mucosa. We also analyzed the enrichment plot of six Gene Ontology (GO) among RNA process, which evidently showed that RNA metabolic processes and RNA modifications such as methylation are most enriched, indicating that the biogenesis and development of CAC may be closely associated with involvement of RNA metabolism and its epigenetics (Figure 3b).
Validation of the Dysregulated mRNAs and lncRNAs in Clinical Samples
To validate the results from our CAC RNA-seq,we chose 8 dysregulated mRNAs and 3 lncRNAs for RTqPCR validation with 30 calcified patient sample pairs (Fig.4). Housekeeping gene GAPDH was used as the endogenous control. The relative expression level of RNAs in patients or control was confirmed to follow normal distribution.In the mRNA group, CAV1, CCDC152, MEST, TSPAN13 were significantly upregulated in blood of CAC patients compared to the normal controls; GMPR, MKRN1,TNS1 and YBX3 were significantly downregulated compared to the normal controls (Figure 4a). In the lncRNA group, AL133477.2 was significantly upregulated in CAC patients,while AL136526.1 and AC007686.4 were significantly downregulated in CAC patients, compared to the normal controls (Figure 4b). The present results showed that the RT-qPCR verification was in accordance with RNA-seq results with high confidence.
Analysis of the Dysregulated circRNAs and miRNAs
CircRNAs and miRNAs, as well as their functional interactions, play vital roles in the regulation of many biological processes including CAC. We then set out to analyze the differentially expressed circRNAs and miRNAs with filtration criteria (fold changes ≥2.0 and p-values ≤0.05). Hierarchical clustering heatmap was performed to evaluate these RNAs, in which CAC and control samples were respectively classified into different branches (Figure 5a,b). Total ly 22 miRNAs and 39 circRNAs were differentially expressed, with 11 miRNAs (50%), and 28 circRNAs (71.79%) upregulated, while 11 miRNAs (50%), and 11 circRNAs (28.21%) were downregulated. For circRNAs, no significant difference in the overall circRNA expression was observed (Figure S1a,b), however, we noticed that the spliced length of the these circRNAs in CAC group is significantly shorter than the control group, which might also account for a significant feature of CAC patients, which need further studies (Figure S1c,d).
To validate the differentially expressed circRNAs from CAC RNA-seq, we chose 6 dysregulated circRNAs for RT-qPCR validation with 30 calcified patient sample pairs (Figure 5c). Housekeeping gene GAPDH was used as the endogenous control. CircDTL, circHAGH, circLZIC, circMCU,circPHF7 and circTOP1 were significantly upregulated in blood of patients with CAC compared to the normal control,which were all in accordance with RNA-seq results with high confidence.
Construction of circRNA-miRNA-mRNA Network of CAC
CircRNAs participate in a variety of biological processes through multiple mechanisms due to their unique structures and properties. We then investigated the above 6 validated circRNAs based on their structures and predicted their functional mechanisms (Figure 6a). Three criteria for circRNAs were mainly focused: MRE (MicroRNA Element) for potential miRNA binding capabilities; RBP (RNA Binding Protein) for circRNA-RBP interaction; ORF (Open Reading Frame) for circRNA as a potentially translatable template. All the circRNAs examined have predicted RBP binding sites, suggesting they might function as RNA-binding protein scaffold. CircDTL, circHAGH and circTOP1 have ORF which indicated that they possess capability for translation and may serve as functional polypeptides. CircRNAs with miRNA binding sites could bind directly to the corresponding miRNAs to inhibit miRNA activity and thus regulate the expression of target genes. All the 6 circRNAs have miRNA binding sites, indicating they might function through miRNA sponging, which is also the most reported circRNA function. In order to investigate the potential function for circRNAs acting as miRNA sponges in CAC, we hence constructed circRNA-miRNA-mRNA network based on our validated 6 circRNAs, and their corresponding regulating miRNAs and mRNAs in CAC. 16 miRNAs, and 31 mRNAs were found to be correlated with this ceRNA network (Figure 6b).
It is known that circRNAs could serve as miRNA sponges to regulate gene expression in different diseases,and these circRNA-miRNA interactions are mediated by the RNA-induced silencing complex (RISC) containing Argonaute2 (AGO2) and many associated proteins. We thus conducted AGO2 RIP assay to determine whether these validated circRNAs in this study could function as miRNA sponges (Figure 6c). Only circTOP1 exhibited strong binding capability towards AGO2, indicating functionally circ-TOP1 might interact with one or multiple miRNAs in CAC.On the other hand, the rest of the circRNAs examined showed no binding with AGO2, which revealed that miRNA sponge mechanistically might not be a common feature for most circRNAs despite multiple predicted miRNA binding sites, at least for the circRNAs examined in this study.
Discussion
Coronary artery disease is a significant cause of morbidity and mortality and the leading cause of sudden cardiac death in adults [48]. Coronary artery calcification, a critical feature of coronary artery disease, is universally found in coronary artery disease patients, especially those with severe symptoms. CAC was previously thought to be a benign process that increases with aging [6]. Subsequent studies suggest that CAC associates with arterial stiffness,which increases risk for adverse cardiovascular events [49].Notably, CAC is positively associated with the degree of atherosclerosis and future cardiac events, and the high prevalence of CAC in coronary heart disease patients makes percutaneous coronary intervention (PCI) difficult to perform [6,50]. As CAC could be hardly detectable during the early stage of atherosclerosis, it calls for early and fast means of prognosis and diagnosis for CAC.
High-throughput sequencing technologies have facilitated the detection of aberrantly expressed both protein coding and noncoding genes in human at transcriptome level. The number of studies focusing on the disease-related RNAs is fast increasing as differential expression of specific genes would positively or negatively correlate with disease pathology. Increasing evidence long suggest that lncRNAs,miRNAs, and circRNAs along with mRNAs could massively involve in the progression of many diseases, with some of them being identified as potentially suitable biomarkers.The pervasiveness of the bloodstream and its perfusion through all organs and tissues enables specific noncoding RNAs to be distributed and function throughout the circulation.Lines of evidence suggest that circulating noncoding RNAs are promising molecules to serve as a minimally invasive diagnostic and prognostic biomarker for various types of cardiovascular disease. The applications of these RNAs often provides critical information to identify and categorize patients toward an individual risk profile; monitor the conditions of disease dynamically, and offer effective prognosis for patients with appropriate treatments. Besides, the circulating noncoding RNAs exhibit tissue- and disease-specific expression patterns, as well as their high stability in the circulatory system, which make them suitable molecules for targeted therapy [51]. Recent advances for the development of noncoding RNA-based targeted strategy are mostly coming from cancer research, with multiple clinical trials are ongoing.For instance, PCA3 (prostate cancer antigen 3) is one of the first noncoding RNA biomarkers in the treatment of prostate cancer [52]. It remains to be seen in the next decade how this field would develop with more precising sequencing technologies and detection measures to validate the feasibility of noncoding RNAs.
However, there is only a limited number of studies for the universal investigations on the dysregulated RNAs of CAC, especially circRNAs. In this study, we utilized RNA deep-sequencing from peripheral blood of CAC patients,and totally identified 238 mRNAs, 155 lncRNAs, 22 miRNAs and 39 circRNAs that were differentially expressed. To the best of our knowledge, it is the first comprehensive study that identifies and analyzes the dysregulated mRNA,miRNA, lncRNA and circRNA in CAC. We also validated some of the dysregulated RNAs which was in accordance with the RNA-seq with high confidence. We also performed GSEA analysis with either upregulated or downregulated differentially expressed RNAs. Several critical pathways that associate with CAC were found in the analysis, especially RNA modifications and RNA metabolism [53,54], both of which are strongly correlated with progression of coronary artery disease, including CAC. m6A-SNPs were found to be associated with coronary artery diseases, in which SNP rs12286 was significantly associated with coronary artery disease at genome-wide level and might regulate the expression of ADAMTS7 [55]. Besides, m6A also participates in vascular calcification, and study on both cell and rat models of calcification reveals that METTL14-dependent m6A plays a vital role in the underlying pathomechanisms [56].Studies also indicated that metabolic syndrome (MS) was correlated with an increased risk of CAC [57]. Further studies are worthy of investigating whether the dysregulated RNAs function in these biological processes.
lncRNA-mRNA interaction is one of the most frequent functional mechanisms of lncRNA [23]. We also established lncRNA-mRNA co-expression network in CAC,based on ceRNA mechanism which is similar to circRNA-miRNA interaction. It is long acknowledged that RNAseq-based lncRNA-mRNA network has become a useful tool to predict functional lncRNAs and their potential functional mechanisms, which is widely used in many disease models and clinical samples [58,59]. Our lncRNA-mRNA network in CAC would hopefully provide useful information to the future CAC investigations.
Lines of evidence reveal that circRNAs participate in various biological processes, including cell proliferation,protein metabolism, autophagy, tumor immunology, signal transduction, genome stability, etc. [31,60]. In this study,we presented 6 circRNAs that were also validated in the subsequent experiments, and we demonstrated their potential mechanisms according to their nucleotide sequences. CircRNAs are generally found to function with multiple mechanisms, such as miRNA sponge, protein scaffold, transcriptional regulation and translation [31,60]. Of note, circRNA acting as miRNA sponge was first found in 2013 and has become one of the most investigated mechanisms in almost all disease models [41,61]. It is believed that circRNA, which harbors miRNA binding sites, would sequester miRNA(s) like a sponge and thus alleviate miRNA’s suppression of its target protein(s). Based on this, we first constructed the circRNA-miRNA-mRNA network in CAC, and 6 circRNAs, 7miRNAs, 116 mRNAs were potentially correlated and were involved in the occurrence and progression of CAC, which indicates potential biological or clinical mechanisms for future studies. AGO2 RIP further indicated that circTOP1 might function as miRNA sponge, whereas the rest circRNA examined in this study showed no significant binding capability towards AGO2. This finding suggests that circRNA may, but not generally, function as sponge for miRNA, at least in this study. However, there is certain possibility that these circRNAs might functionally interact with miRNAs in other contexts due to the cell- and tissue- specificity of circRNAs. Together with lncRNA-mRNA co-expression network, we believe that a comprehensive understanding of the complex networks of interactions that these dysregulated RNAs would provide a unique opportunity for better therapeutic interventions
However, some further questions remain that require future investigations. First in our preliminary design, we aimed to choose patients in both groups in which none of them has chronic diseases such as hypertension, diabetes,and chronic renal insufficiency, which narrowed our enrollment for patients. Despite some previous work used comparable number of sample pairs for RNA-seq [68-71], more CAC samples are still expected for the RNA-seq and analysis based on sample size estimation, which could help us more optimize the study design for differential expression detection with high confidence. Second, it should be noted that we used peripheral blood from CAC patients for RNAseq rather than calcified lesion on the coronary artery due to lack of these samples in clinical practices. It would be beneficial to include calcified lesion samples from coronary arteries in future studies to provide a more direct assessment of the disease process. Generally, diagnostic value of the established biomarkers may be limited by the heterogeneity of many factors of patients. Addition of calcified lesion from a larger cohort of patients would provide more accurate landscape of CAC. Third, the RNA regulatory networks are only based on bioinformatics predictions and lack practical experiment for verification which requires future in-depth studies with in vivo and in vitro experiments. Besides,use of animal models facilitates the investigations in cardiovascular diseases [72,73], therefore these models would also help provide a systematic view on the potential functions and evolutionary conservation of the RNAs during CAC. To investigate further the conserved functions of the differentially expressed RNAs, we are currently committed to studying these potential mechanisms with CAC mouse models.
Conclusion
In this study, we have comprehensively sequenced and analyzed the dysregulated mRNAs, lncRNAs, miRNAs and circRNAs in peripheral blood of CAC patients, with some of the RNAs validated in clinical samples. Also, we constructed the lncRNA-mRNA co-expression network,and for the first time established the circRNA-miRNA-mRNA in CAC. GSEA pathway analysis indicated the progression of CAC might correlate with RNA modifications and RNA metabolism, which provides better insights for the future studies in the CAD research. All the present results provide valuable resources for the future clinical prognosis and predictions of CAC.
Declarations
Ethical Approval and Consent to Participate
The study was approved by the Human Research Ethics Committee of University of Science and Technology of China (2019KY ER No.165).Written informed consent was obtained from each patient for this study. All methods were carried out in accordance with relevant guidelines and regulations (Declaration of Helsinki).
Disclosure Statement
The authors report there are no competing interests to declare.
Author Contribution
L.M., L.C., G.S. and H.H. conceived and initiated this research. L.C., L.M., G.S. and H.H. designed the project,provided the major funding, and supervised the experiments.H.H., L.C., Y.W. and Z.W. performed the experiments and analyzed the data. Y.W. performed the bioinformatics analysis. L.C. and H.H. wrote the manuscript. All authors have discussed the results and made comments on the manuscript. All authors approved the final manuscript.
Acknowledgements
We appreciate the support of patients in this study.
Funding
The study was supported by the National Natural Science Foundation of China (81870192, 82170263), the National Key R&D Program of China (2019YFA0802600), and Anhui Health Commission Research Program (AHWJ2021b080)
- Chen J, Budoff MJ, Reilly MP, Yang W, Rosas SE et al.(2017) Coronary Artery Calcification and Risk of Cardiovascular Disease and Death Among Patients With Chronic Kidney Disease. JAMA Cardiol 2: 635-43.
- Kugathasan P, Johansen MB, Jensen MB, Aagaard J,Nielsen RE, Jensen SE (2019) Coronary Artery Calcification and Mortality Risk in Patients With Severe Mental Illness.Circ Cardiovasc Imaging 12: e008236.
- Wexler L, Brundage B, Crouse J, Detrano R, Fuster Vet al. (1996) Coronary artery calcification: pathophysiology,epidemiology, imaging methods, and clinical implications. A statement for health professionals from the American Heart Association. Writing Group. Circulation 94: 1175-92.
- DeFina LF, Radford NB, Barlow CE, Willis BL, Leonard D et al. (2019) Association of All-Cause and Cardiovascular Mortality With High Levels of Physical Activity and Concurrent Coronary Artery Calcification. JAMA Cardiol 4:174-81.
- Agarwal S, Morgan T, Herrington DM, Xu J, Cox AJ etal. (2011) Coronary calcium score and prediction of all-cause mortality in diabetes: the diabetes heart study. Diabetes Care 34: 1219-24.
- Liu W, Zhang Y, Yu CM, Ji QW, Cai M et al. (2015) Current understanding of coronary artery calcification. J Geriatr Cardiol 12: 668-75.
- Peng ALW, Dardari ZA, Blumenthal RS, Dzaye O,Obisesan OH et al. (2021) Very High Coronary Artery Calcium (>= 1000) and Association With Cardiovascular Disease Events, Non-Cardiovascular Disease Outcomes, and Mortality Results From MESA. Circulation 143: 1571-83.
- Chen CC, Hsieh IC, Liu YC, Chan T, Wen MS, Wan YL (2011) The effect of calcium score on the diagnostic accuracy of coronary computed tomography angiography. Int J Cardiovasc Imaging 27: 37-42.
- Budoff MJ, Achenbach S, Blumenthal RS, Carr JJ,Goldin JG et al. (2006) Assessment of coronary artery disease by cardiac computed tomography: a scientific statement from the American Heart Association Committee on Cardiovascular Imaging and Intervention, Council on Cardiovascular Radiology and Intervention, and Committee on Cardiac Imaging,Council on Clinical Cardiology. Circulation 114:1761-91.
- Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR,Viamonte M et al. (1990) Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol 15: 827-32.
- Goodall GJ, Wickramasinghe VO (2021) RNA in cancer.Nature Reviews Cancer 21: 22-36.
- Salta E, De Strooper B (2017) Noncoding RNAs in neurodegeneration. Nat Rev Neurosci 18: 627-40.
- Das S, Shah R, Dimmeler S, Freedman JE, Holley C etal. (2020) Noncoding RNAs in Cardiovascular Disease: Current Knowledge, Tools and Technologies for Investigation,and Future Directions: A Scientific Statement From the American Heart Association. Circ Genom Precis Med 13: e000062.
- O'Donnell CJ, Kavousi M, Smith AV, Kardia SLR, Feitosa MF et al. (2011) Genome-Wide Association Study for Coronary Artery Calcification With Follow-Up in Myocardial Infarction. Circulation 124: 2855-255.
- Cheng J, Wen J, Wang N, Wang C, Xu QB, Yang Y (2019) Ion Channels and Vascular Diseases. Arteriosclerosis Thrombosis and Vascular Biology 39: 146-56.
- Li JJ, Zhu CG, Yu B, Liu YX, Yu MY (2007) The role of inflammation in coronary artery calcification. Ageing Research Reviews 6: 263-70.
- Rodriguez-Sinovas A, Sanchez JA, Valls-Lacalle L,Consegal M, Ferreira-Gonzalez I (2021) Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease.International Journal of Molecular Sciences 22.
- Sen SK, Boelte KC, Barb JJ, Joehanes R, Zhao X et al. (2014) Integrative DNA, RNA, and protein evidence connects TREML4 to coronary artery calcification. Am J Hum Genet 95: 66-76.
- Sen SK, Barb JJ, Cherukuri PF, Accame DS, Elkahloun AG et al. (2014) Identification of candidate genes involved in coronary artery calcification by transcriptome sequencing of cell lines. BMC Genomics 15: 198.
- O'Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of MicroRNA Biogenesis, Mechanisms of Actions,and Circulation. Frontiers in endocrinology 9: 402.
- Liu J, Xiao X, Shen Y, Chen L, Xu C et al. (2017) MicroRNA-32 promotes calcification in vascular smooth muscle cells: Implications as a novel marker for coronary artery calcification.PLoS ONE 12: e0174138.
- Liu W, Ling S, Sun W, Liu T, Li Y et al. (2015) Circulating microRNAs correlated with the level of coronary artery calcification in symptomatic patients. Sci Rep 5: 16099.
- Statello L, Guo CJ, Chen LL, Huarte M (2021) Gene regulation by long non-coding RNAs and its biological functions.Nat Rev Mol Cell Biol 22: 96-118.
- Sallam T, Sandhu J, Tontonoz P (2014) Long Noncoding RNA Discovery in Cardiovascular Disease: Decoding Form to Function. Circ Res 2018, 122(1):155-166.
- Vausort M, Wagner DR, Devaux Y: Long noncoding RNAs in patients with acute myocardial infarction. Circ Res 115: 668-77.
- Kumarswamy R, Bauters C, Volkmann I, Maury F,Fetisch J (2014) Circulating long noncoding RNA, LIPCAR,predicts survival in patients with heart failure. Circ Res 114:1569-75.
- Pierce JB, Feinberg MW (2020) Long Noncoding RNAs in Atherosclerosis and Vascular Injury: Pathobiology,Biomarkers, and Targets for Therapy. Arterioscler Thromb Vasc Biol 40: 2002-17.
- Jeong G, Kwon DH, Shin S, Choe N, Ryu J (2019) Long noncoding RNAs in vascular smooth muscle cells regulate vascular calcification. Sci Rep 9: 5848.
- Cai Y, Yang YJ, Chen XW, Wu GZ, Zhang XQ et al.(2016) Circulating 'lncRNA OTTHUMT00000387022' from monocytes as a novel biomarker for coronary artery disease.Cardiovasc Res 112: 714-24.
- Zhang L, Zhang Y, Wang Y, Zhao YF, Ding H et al.(2020) Circular RNAs: Functions and Clinical Significance in Cardiovascular Disease. Front Cell Dev Biol 8.
- Chen L, Shan G (2021) CircRNA in cancer: Fundamental mechanism and clinical potential. Cancer Lett 505:49-57.
- Verduci L, Tarcitano E, Strano S, Yarden Y, Blandino G (2021) CircRNAs: role in human diseases and potential use as biomarkers. Cell Death & Disease 12.
- Wang LY, Shen C, Wang YY, Zou TY, Zhu HJ et al.(2019) Identification of circular RNA Hsa_circ_0001879 and Hsa_circ_0004104 as novel biomarkers for coronary artery disease. Atherosclerosis 286: 88-96.
- Gao Y, Liu Y, Fu Y, Wang Q, Liu Z et al. (2021) The potential regulatory role of hsa_circ_0004104 in the persistency of atrial fibrillation by promoting cardiac fibrosis via TGF--beta pathway. BMC cardiovascular disorders 21: 25.
- Wang K, Long B, Liu F, Wang JX, Liu CY et al.(2016) A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J.
- Du WW, Yang W, Chen Y, Wu ZK, Foster FS et al.(1990) Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur Heart J.
- Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR,Viamonte M (1990) Quantification of Coronary-Artery Calcium Using Ultrafast Computed-Tomography. J Am Coll Cardiol 15: 827-32.
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37: 907-15.
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923-30.
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550.
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger Jet al. (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333-8.
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10.
- Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife 4.
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JTet al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498-504.
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL et al. (2005) Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102: 15545-50.
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S et al. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267-73.
- Liu X, Wang X, Li J, Hu S, Deng Y et al. (2020) Identification of mecciRNAs and their roles in the mitochondrial entry of proteins. Sci China Life Sci.
- Vahatalo J, Holmstrom L, Pakanen L, Kaikkonen K,Perkiomaki J et al. (2021) Coronary Artery Disease as the Cause of Sudden Cardiac Death Among Victims < 50 Years of Age. AmJC 147: 33-8.
- Guo JC, Fujiyoshi A, Willcox B, Choo J, Vishnu A etal. (2017) Increased Aortic Calcification Is Associated With Arterial Stiffness Progression in Multiethnic Middle-Aged Men. Hypertension 69: 102-8.
- Criqui MH, Knox JB, Denenberg JO, Forbang NI, Mc-Clelland RL et al. (2017) Coronary Artery Calcium Volume and Density Potential Interactions and Overall Predictive Value:The Multi-Ethnic Study of Atherosclerosis. Jacc-Cardiovasc Imag 10: 845-54.
- Quinn JJ, Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17:47-62.
- Groskopf J, Aubin SM, Deras IL, Blase A, Bodrug S etal. (2006) APTIMA PCA3 molecular urine test: development of a method to aid in the diagnosis of prostate cancer. Clin Chem 52: 1089-95.
- Dorn LE, Tual-Chalot S, Stellos K, Accornero F (2019) RNA epigenetics and cardiovascular diseases. J Mol Cell Cardiol 129: 272-80.
- Chang Y, Kim BK, Yun KE, Cho J, Zhang YY et al.(2014) Metabolically-Healthy Obesity and Coronary Artery Calcification. J Am Coll Cardiol 63: 2679-86.
- Mo XB, Lei SF, Zhang YH, Zhang H (2018) Detection of m(6)A-associated SNPs as potential functional variants for coronary artery disease. Epigenomics 10:1279-87.
- Chen J, Ning YC, Zhang H, Song NN, Gu YL et al.(2019) METTL14-dependent m6A regulates vascular calcification induced by indoxyl sulfate. Life Sci 239.
- Adeseun GA, Rivera ME, Thota S, Joffe M, Rosas SE (2008) Metabolic syndrome and coronary artery calcification in renal transplant recipients. Transplantation 86: 728-32.
- Wang W, Gao FL, Zhao Z, Wang HY, Zhang L et al.(2017) Integrated Analysis of LncRNA-mRNA Co-Expression Profiles in Patients with Moyamoya Disease. Sci Rep-Uk 7.
- Sun YX, Li C, Lu QY, Jiang HX, Zhu MM et al.(2021) Integrative Analysis of lncRNA-mRNA Profile Reveals Potential Predictors for SAPHO Syndrome. Frontiers in Genetics 12.
- Chen L, Huang C, Wang XL, Shan G (2015) Circular RNAs in Eukaryotic Cells. Curr Genomics 16: 312-8.
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495: 384-8.
- Wang J, Zhang C (2021) Identification and validation of potential mRNA- microRNA- long-noncoding RNA (mRNA-miRNA-lncRNA) prognostic signature for cervical cancer.Bioengineered 12: 898-913.
- Yang L, Lu P, Yang X, Li K, Chen X, Qu S (2021) Excavating novel diagnostic and prognostic long non-coding RNAs (lncRNAs) for head and neck squamous cell carcinoma:an integrated bioinformatics analysis of competing endogenous RNAs (ceRNAs) and gene co-expression networks.Bioengineered 12: 12821-38.
- Chen B, Deng Y, Wang B, Tian Z, Tong J et al. (2021)Integrated analysis of long non-coding RNA-microRNA-mRNA competing endogenous RNAregulatory networks in thromboangiitis obliterans. Bioengineered 12: 12023-37.
- Zou D, Wang Y, Wang M, Zhao B, Hu F et al. (2021) Bioinformatics analysis reveals the competing endogenous RNA (ceRNA) coexpression network in the tumor microenvironment and prognostic biomarkers in soft tissue sarcomas.Bioengineered 12: 496-506.
- Zhang Q, Sun YG, Wang C, Shao F (2022) Circular RNA-microRNA-mRNA network identified circ_0007618 and circ_0029426 as new valuable biomarkers for lung adenocarcinoma.Bioengineered 13: 6258-71.
- Li ZM, Sun Y, He ML, Liu JW (2021) Differentially--expressed mRNAs, microRNAs and long noncoding RNAs inintervertebral disc degeneration identified by RNA-sequencing.Bioengineered 12: 1026-39.
- Ji WF, Chen JX, He S, Zhou YQ, Hua L et al. (2021) Characteristics of circular RNAs expression of peripheral blood mononuclear cells in humans with coronary artery disease.Physiol Genomics 53: 349-57.
- Kong FC, Lv ZY, Wang LF, Zhang K, Cai Y et al. (2021) RNA-sequencing of peripheral blood circular RNAs in Parkinson disease. Medicine (Baltimore) 100.
- Sun Y, Zhang D, Sun G, Lv Y, Li Y et al. (2018) RNAsequencing study of peripheral blood mononuclear cells in sporadic Meniere's disease patients: possible contribution of immunologic dysfunction to the development of this disorder.Clin Exp Immunol 192: 33-45.
- Sheng Z, Wang X, Xu G, Shan G, Chen L (2019) Analyses of a Panel of Transcripts Identified From a Small Sample Size and Construction of RNA Networks in Hepatocellular Carcinoma. Front Genet 10: 431.
- Jia T, Wang C, Han ZX, Wang XZ, Ding M, Wang QY (2020) Experimental Rodent Models of Cardiovascular Diseases. Front Cardiovasc Med 7.
- Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL,Blanco-Colio L, Lavin B et al. (2011) Animal Models of Cardiovascular Diseases. J Biomed Biotechnol.
FIGURE
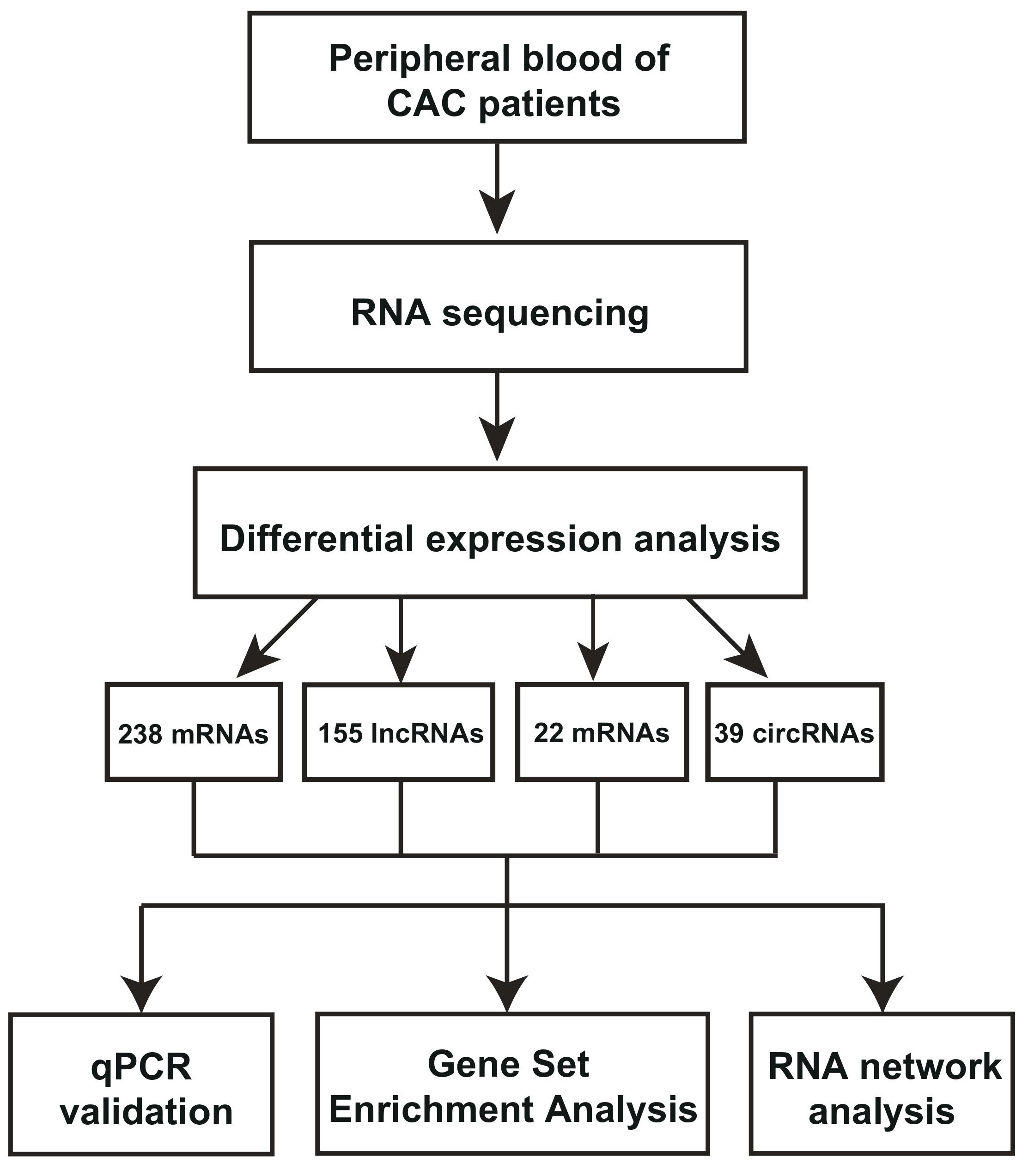
Figure : Graphical abstract
FIGURE 1
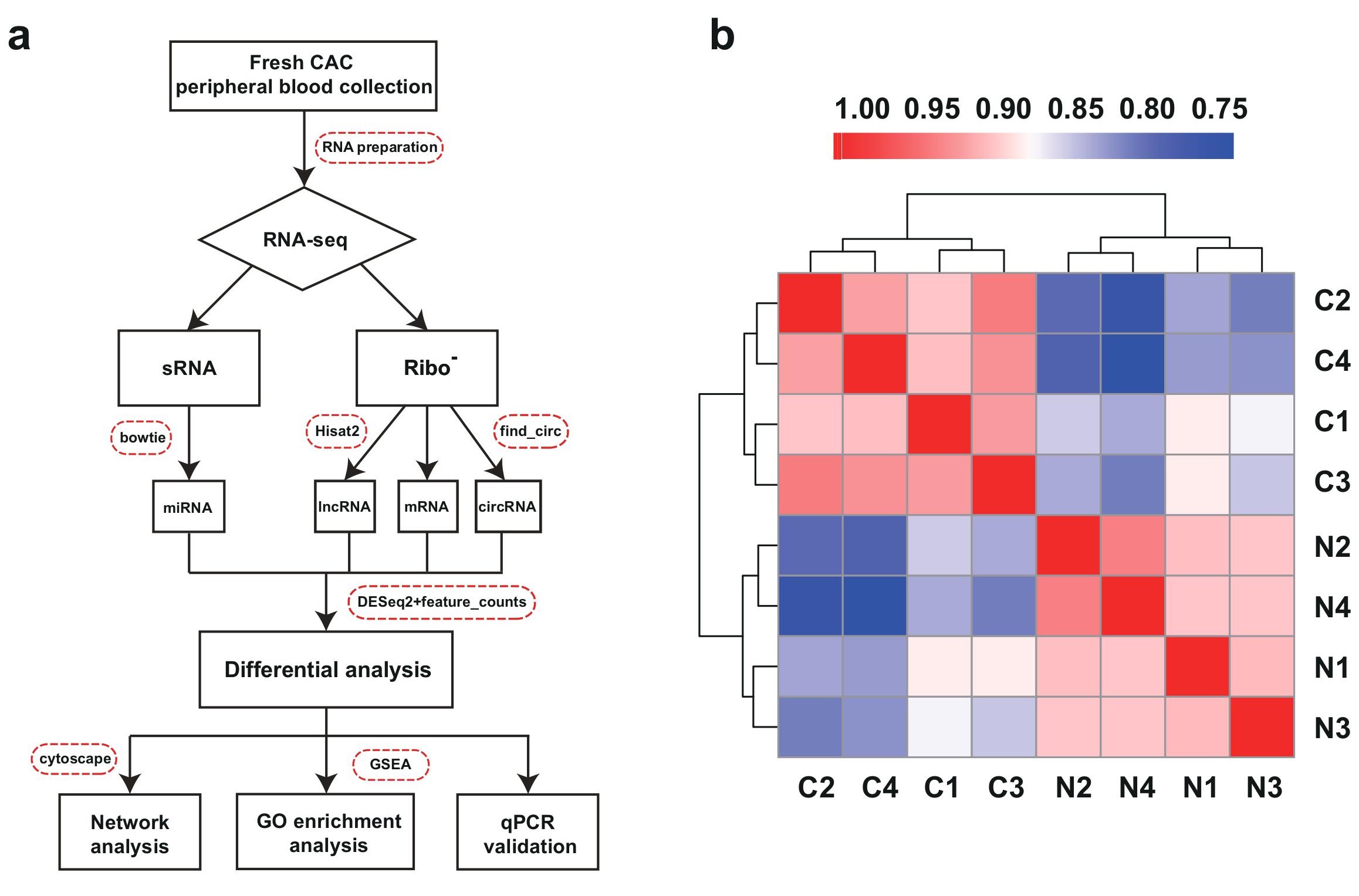
Figure 1: RNA-seq analysis of differentially expressed RNAs from peripheral blood of coronary artery calcification (CAC) patients. a, The workflow of CAC RNA-seq and in-depth investigations in this study. The bioinformatics software were also indicated. b, Heatmap of correlation between calcification (C) or normal samples (N) by Pearson’s correlation coefficient analysis
FIGURE 2
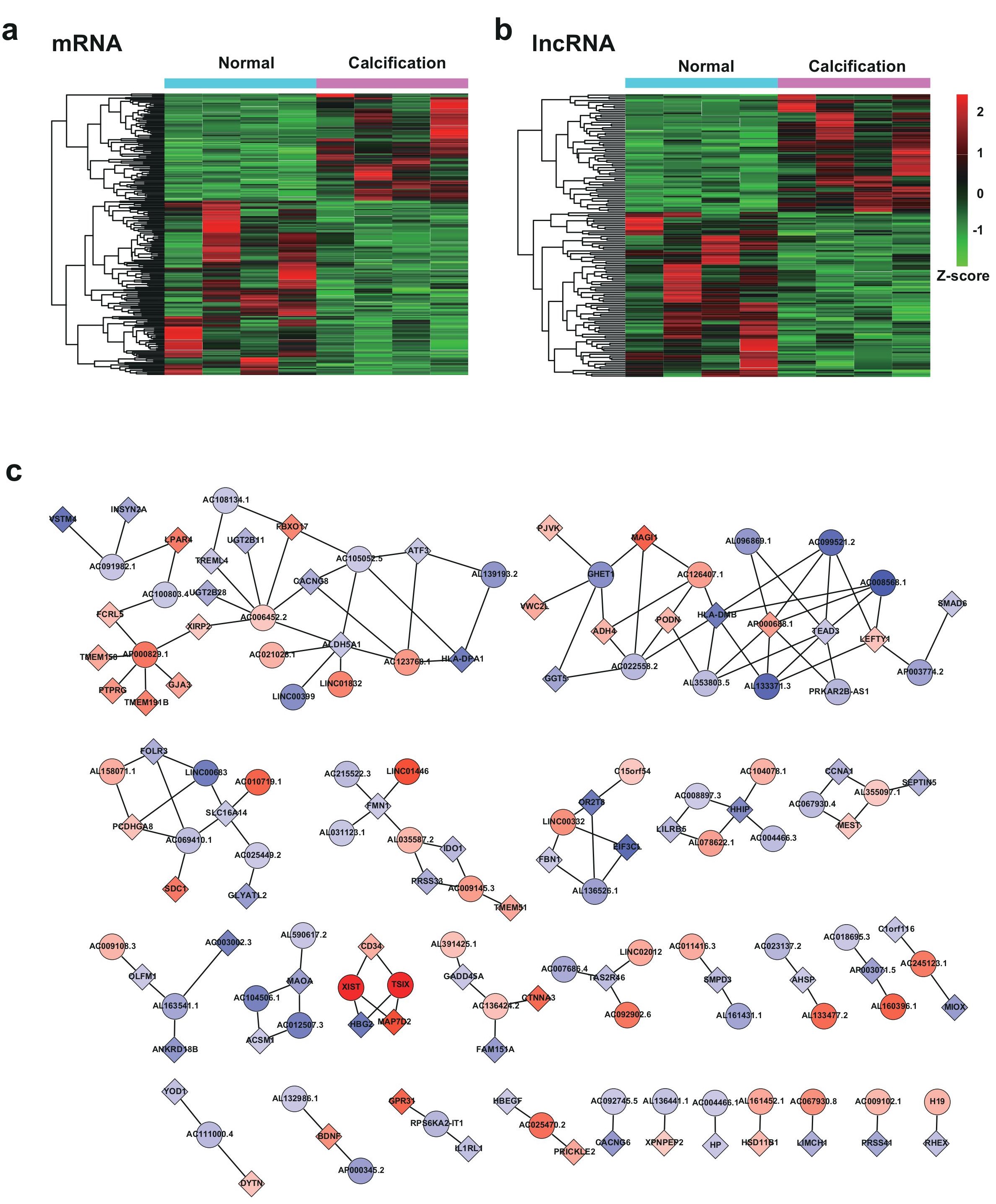
Figure 2: Bioinformatics analysis of differentially expressed mRNAs and lncRNAs in CAC. a, Hierarchical cluster heatmap of the differentially expressed mRNA in CAC. b, Hierarchical cluster heatmap of the differentially expressed lncRNAs in CAC. c, Representation of the lncRNA- mRNA co-expression network (correlation coefficient absolute value >0.99). The boxes represent mRNA, and the circles represent lncRNA. Red, upregulated; blue, downregulated
FIGURE 3
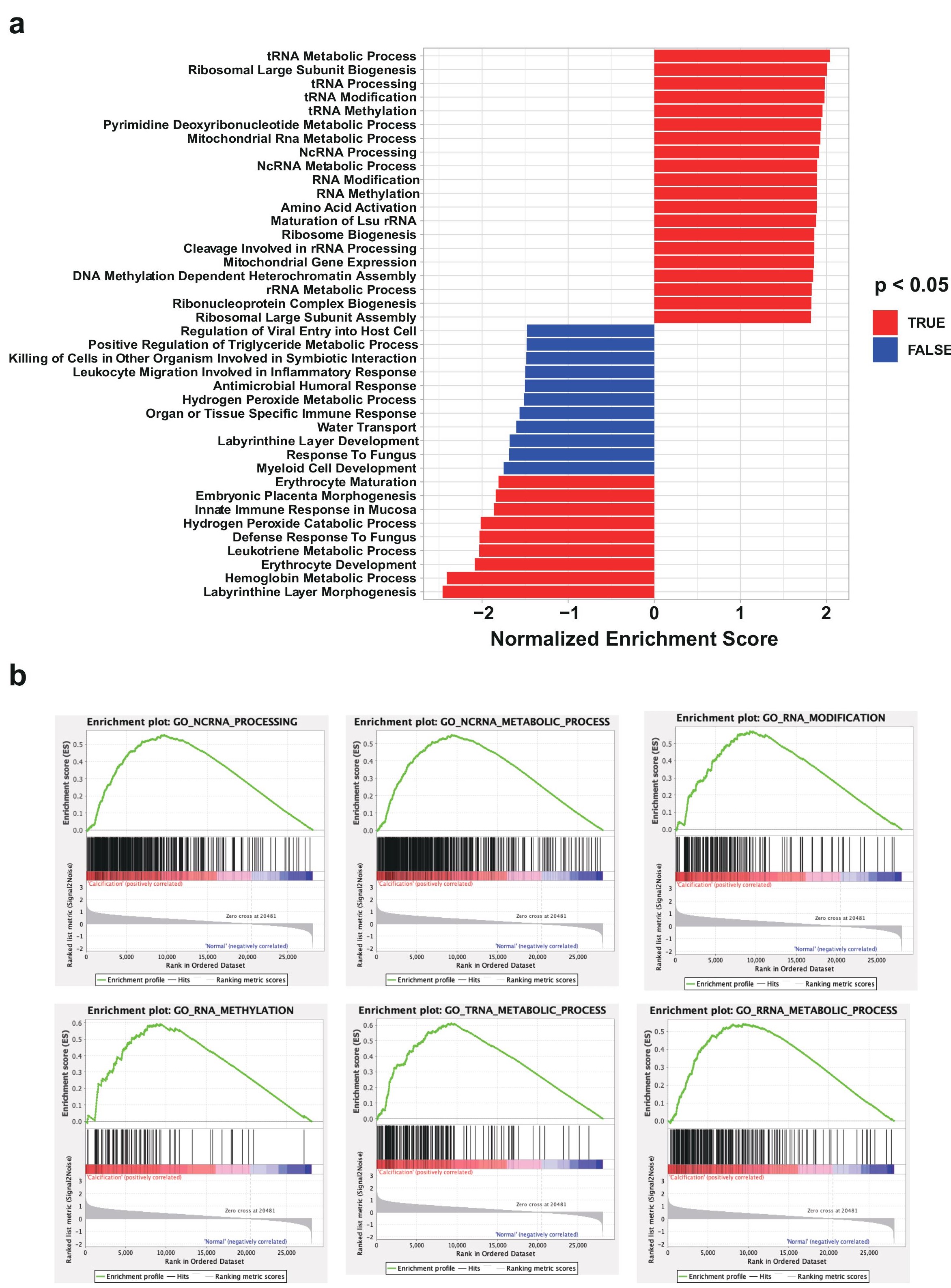
Figure 3: Gene Set Enrichment Analysis (GSEA) analysis of differentially expressed mRNAs in CAC RNA-seq. a, GSEA bar plot of the up-regulated or down-regulated gene ontology (GO) term in CAC. b, Representation of enrichment plot of the six RNA process-related GO ontology
FIGURE 4
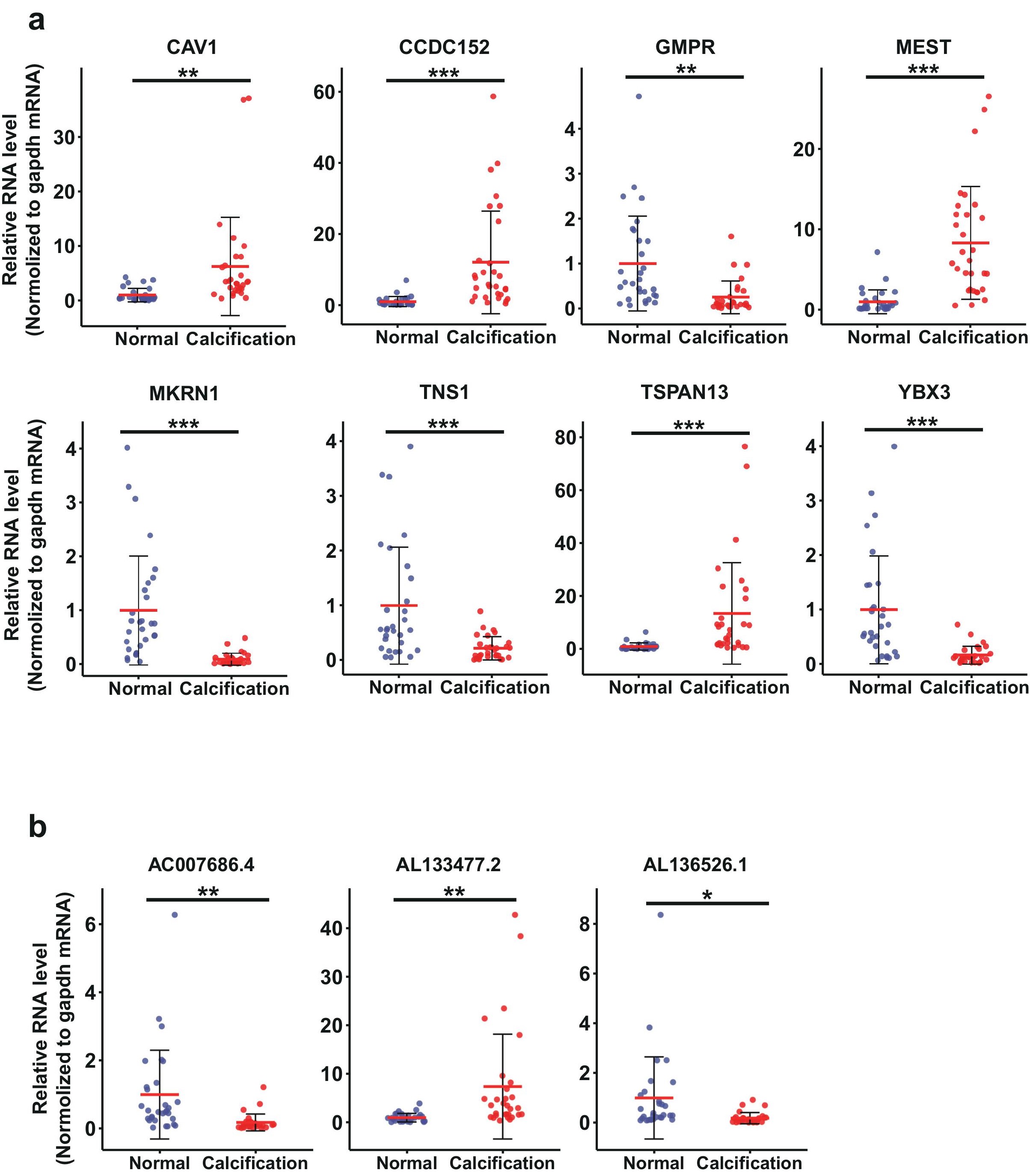
Figure 4: Validation of mRNAs and lncRNAs identified from RNA-seq by RT-qPCR in clinical CAC samples. a, The RT-qPCR validation results of 8 dysregulated mRNAs. b, The RT-qPCR validation results of 3 dysregulated lncRNAs. The patients information of the CAC samples for validation are included in the Table S3. Data are median SD. *p < 0.05, **p < 0.01, ***p < 0.001 are based on the Student’s t-test
FIGURE 5
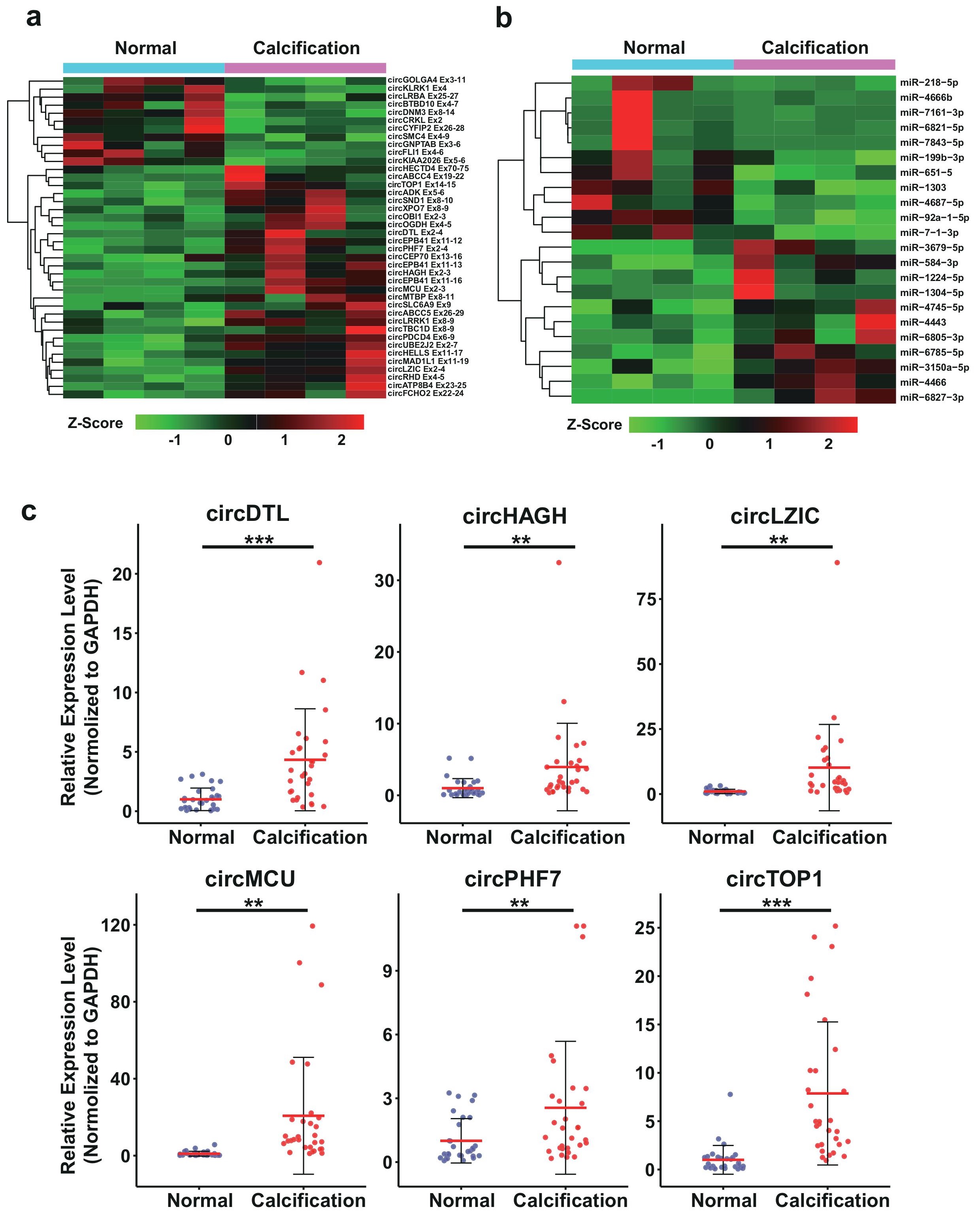
Figure 5: The bioinformatics analysis and RT-qPCR validation of the differentially expressed circRNAs and miRNAs. a, Hierarchical cluster heatmap of differentially expressed circRNA. b, Hierarchical cluster heatmap of differentially expressed miRNA. c, RT-qPCR validation of circRNAs identified from RNA-seq in clinical CAC samples. The patients information of the CAC samples for validation are included in the Table S3. Data are median SD. *p < 0.05, **p < 0.01, ***p < 0.001 are based on the Student’s t-test
FIGURE 6
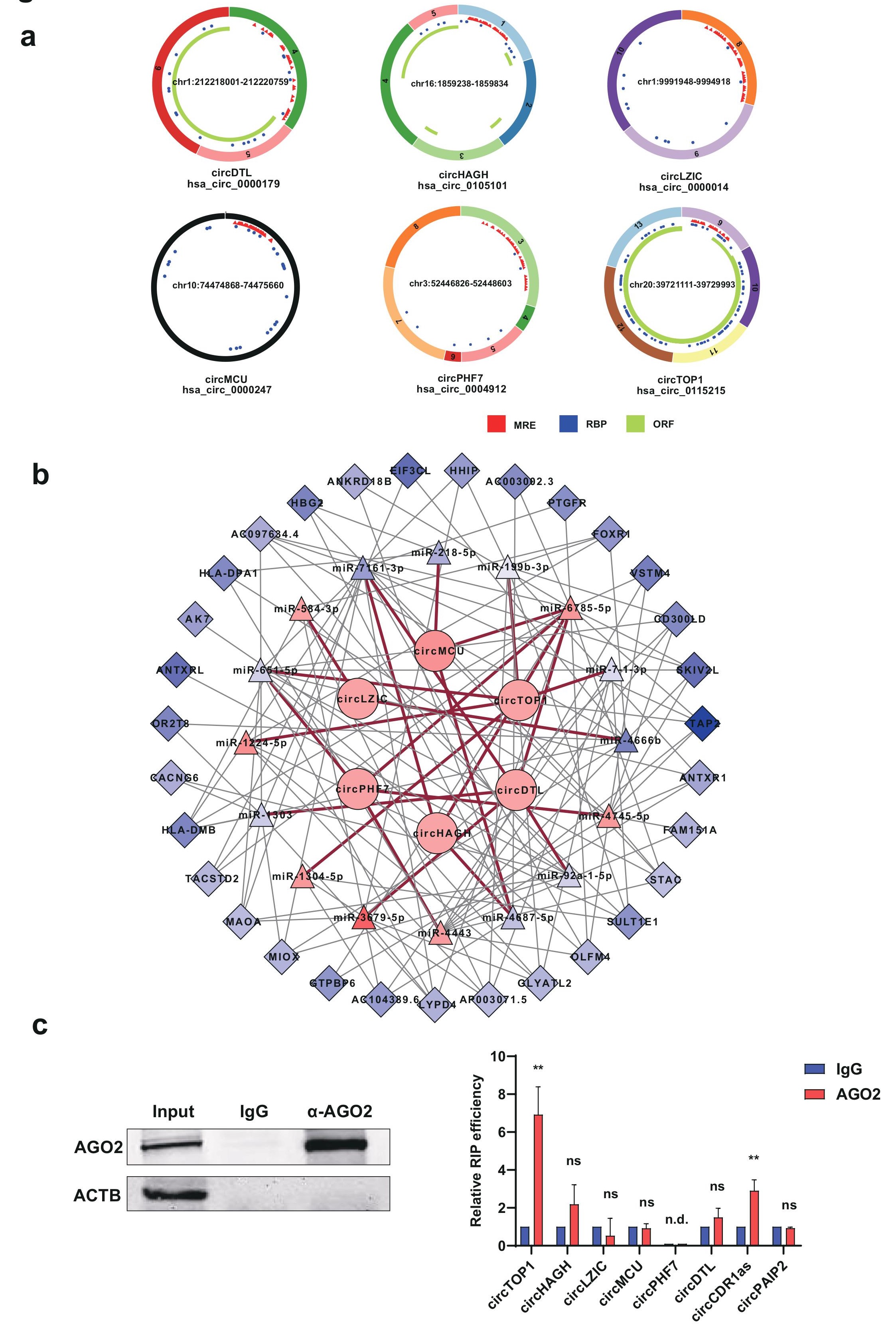
Figure 6: Construction of circRNA-miRNA-mRNA network. a, Predicted functional mechanisms of the validated six circRNAs in the RNAseq. MRE, miRNA response elements. RBP, RNA binding protein. ORF, open reading frame. b, circRNA-miRNA-mRNA interacting network for the validated circRNAs (correlation coefficient absolute value 0.99) . Boxes represent differentially expressed mRNA, triangles represent differentially expressed miRNA, circles represent differentially expressed circRNAs. Red line, circRNA-miRNA interaction; grey line mRNA- miRNA interaction. Pink, upregulated; blue, down-regulated
FIGURE S1
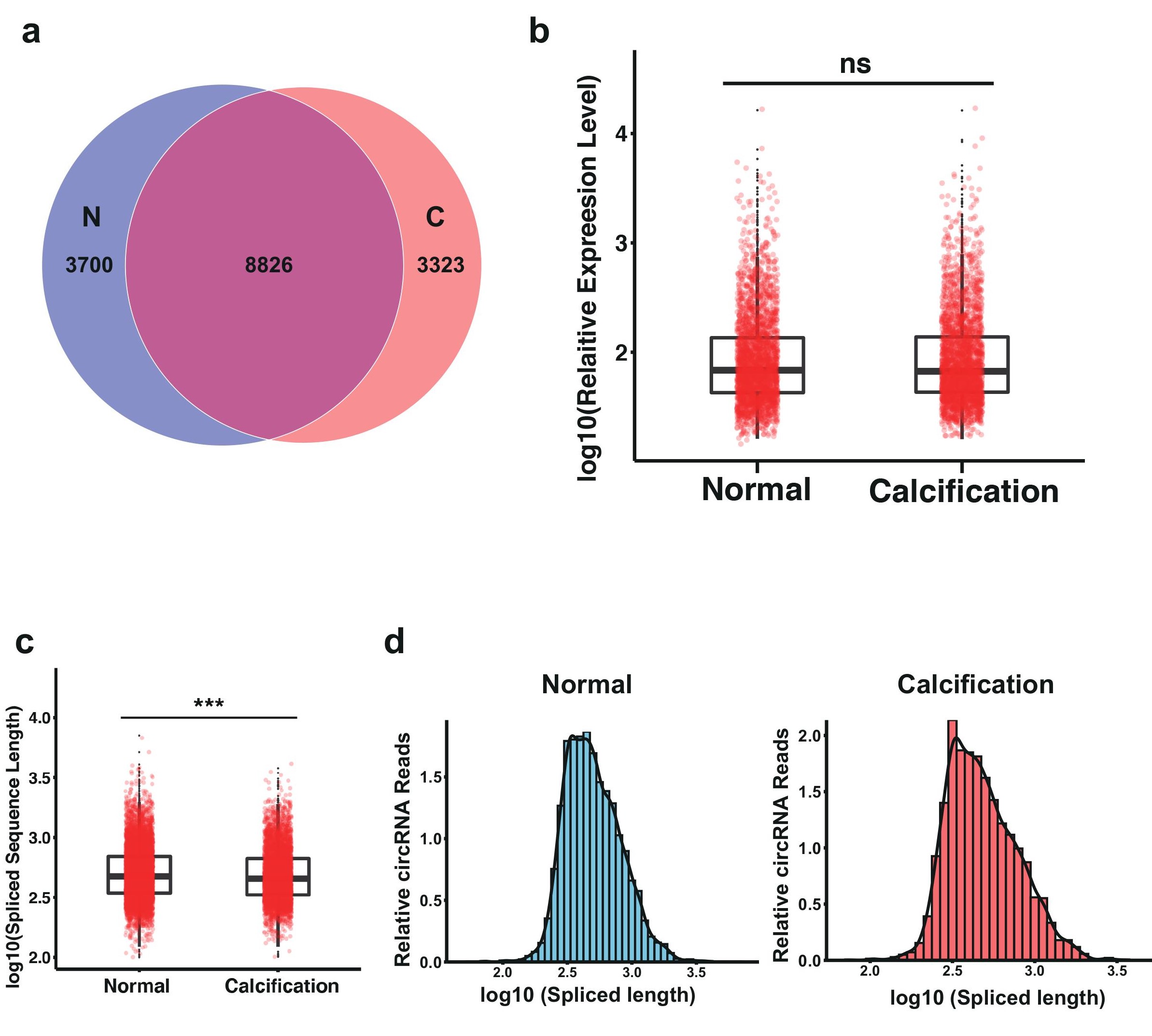
Figure S1: Expression profiling analysis for circRNAs in CAC. (A) Venn plot of the circRNAs identified in CAC and control group. C: CAC; N: normal. (B) Box plot of all circRNAs expression level in CAC and control group.(C) Box plot of all circRNAs spliced sequence length distribution in CAC and control group. (D-E) The distribution of circRNAs spliced sequence length in CAC and control group
Tables at a glance
Figures at a glance