A Rare Case of Mucopolysaccharidosis Type - VI in an Indian Teenage Male
Received Date: January 02, 2026 Accepted Date: January 21, 2026 Published Date: January 28, 2026
doi:10.17303/jgdt.2026.1.101
Citation: Roy D, Sen I, Bhadury S, Bandyopadhyay D (2026) A Rare Case of Mucopolysaccharidosis Type - VI in an Indian Teenage Male. J Gen Disor Thera. 1: 101
Abstract
Mucopolysaccharidosis (MPS) is a group of inherited metabolic storage disorders caused by the absence or malfunction of lysosomal enzymes resulting in progressive accumulation of glycosaminoglycans (GAGs) in multiple tissues giving rise to a variety of clinical manifestations including skeletal, facial, ocular, cardiac, respiratory, neurological, lymphoreticular, dental abnormalities. Characteristic clinical findings, radiological and biochemical assay, specific enzymatic testing and finally genetic studies confirms the diagnosis of specific subtypes of MPS. Enzyme replacement therapy (ERT) remains the only key therapeutic option with different prognosis and outcome. We are here reporting a rare case of MPS type - VI with classical multisystem involvements from sub-Himalayan rural India.
Keywords: Mucopolysaccharidosis; Storage Disorders; Glycosaminoglycans; Lysosomal Enzymes; Genetic Study; Enzyme Replacement Therapy
Introduction
Mucopolysaccharidosis (MPS) is a set of diseases resulting from genetic mutation causing lysosomal enzymatic defect which in turn leads to gradual accumulation of Glycosaminoglycans (GAGs) in different tissues producing a series of diverse symptoms and signs notably skeletal deformities (dysostosis multiplex) like short stature, short neck, broad clavicle, joint abnormalities like joint stiffness and hip deformities, coarse facial changes like frontal bossing, depressed nasal bridge, large tongue, and ocular abnormalities predominantly corneal opacity, recurrent respiratory infections, cardiovascular abnormalities particularly mitral valve diseases (thickening / regurgitation), neurological manifestations including intellectual abnormality, seizure disorder, ventriculomegaly, cognitive and behavioral impairment, dental deformities like malocclusion and poor oral hygiene [1, 2]. Diagnosis of MPS is fundamentally based on clinical features, enzymatic testing and genetic testing along with biochemical, laboratory and radiological investigations as a supporting tool.
MPS has seven subtypes classified based on deficiency of lysosomal enzyme variations. MPS - I (Hurler syndrome) is due to abnormalities of alpha- iduronidase enzyme resulting in excretion of dermatan sulphate and heparan sulphate in urine and the most predominant symptoms and signs include corneal opacity, upper airway obstruction and cardiomyopathy. In MPS - II (Hunter syndrome) there is lack of iduronate-2 sulphate sulphatase enzyme and important clinical findings includes mental retardation, loss of hearing, hepatosplenomegaly, and it is having male predominance and X- linked association. In type - III MPS (Sanfilippo syndrome) is of four subtypes (A, B, C, D) having seizure and mental alterations. MPS - IV (Morquio syndrome) there are predominant joint deformities (laxity / stiffness) due to deficiency of galactosamine - 6 - sulfatase and Beta - galactosidase enzymes. MPS - VII (Sly syndrome) is basically for Beta - glucuronidase enzyme deficiency resulting in mental retardation and cognitive behavioral changes in MPS - IX, the rarest type, is due to hyaluronidase enzyme deficiency. In MPS- VI (Maroteux – Lamy syndrome ), a rare autosomal recessive disorder, results from lysosomal enzyme malfunction because of genetic mutation in aryl sulphatase B (ARSB) leading to inappropriate and incomplete catabolism of CAGs (Chondroitin sulphate and Dermatan sulphate) [3]. The most common clinical manifestations in this subtype essentially includes corneal opacity, skeletal dysplasia (dysostosis multiplex), pulmonary malfunctions, organomegaly notably hepatosplenomegaly, otitis, hearing loss, sleep apnea, facial dysmorphism, hydrocephalus, Mongolian spots, hernia etc but with preserved mental- cognitive- behavioral function and optimal intellectual functions and uneventful growth and developmental milestones in most of the cases.
Clinical manifestations of MPS - VI are typically found in 2nd to 3rd decades of life due slow progression unlike other subtypes. Overall incidence of MPS - VI reported worldwide is 1 per 320000 live births with highest incidence found in Eastern Saudi Arabia and North- east Brazil whereas lowest in Poland and South Korea. The incidence is higher in patients born of consanguinity [4].
Case Description
A 14 year Muslim boy from Purnia, Bihar, born from a nonconsanguineous marriage, having one healthy sibling, with no significant birth and family history, who had normal developmental milestones and school performance, developed insidious onset, gradually progressive bowing of his both legs and bending of both hands leading to walking and playing difficulty and facial dysmorphism at the age of 6 years and failure to gain height in next 8 years and whitish discoloration of both eyes for last 4 years, which was progressed to more advanced stage .There was no history of headache, seizure, vomiting, visual and hearing disturbances, palpitation, shortness of breath, recurrent cough and cold, skin rash or pigmentation, bony pain or fracture, weight loss or unusual weight gain, mood swings, etc. On clinical examination there was facial dysmorphism with frontal bossing, depressed nasal bridge, hypertelorism, macrocephaly, coarse thick hair, low set ears, widened thick lips, bilateral corneal clouding, macroglossia, dysostosis multiplex notably genu valgum deformities, broad hands and claw like fingers with flexion deformities of bilateral elbow and wrist , kyphosis , short stature (Upper and lower segment ratio 0.9) hepatosplenomegaly, small umbilical hernia, soft S1, with no respiratory, neurological, lymphoreticular and thyroid abnormalities and normal secondary sexual characters and sexual maturity rating by Tanner staging G4P2 [Fig: 1]. His height was 105.5 cm (< 3rd percentile), height standard deviation score was - 6.4, BMI - 19.3 Kg/m2, CA = B A = 14 years, WA = HA = 5.5 year. His laboratory investigations showed mild anemia, low serum Vitamin- D3, borderline low IGF-I [Fig: 2]. Radiological imaging showed features of Rickets, genu valgum deformity, frontal bossing, J- shaped sella, ventriculomegaly [Fig: 3]. 2-D echocardiography revealed thickened both mitral leaflets, aortic and tricuspid valves and mild mitral stenosis. His urinary was positive for glycoseaminoglycans. Genetic study (next generation sequencing) revealed homozygous missense mutation in ARSB gene (c.1350G>C;p.Trp450Cys) [Fig: 4] and thus confirmed diagnosis of Mucopolysaccharidosis Type VI (Maroteux - Lamy syndrome). Initiated supportive treatment including physiotherapeutic rehabilitation, vitamin D3 supplementation, and Ophthalmology and orthopedic consultation. Enzyme replacement therapy (ERT) with Galsulphase (Naglazyme) is planned for next level of definitive treatment.
Discussion
MPS is an extremely rare, genetically triggered metabolic disorders with diverse clinical features, biochemical alterations from enzymatic malfunctions [Fig: 5] [5]. Type VI MPS is an autosomal recessive disorder with distinct clinical manifestations involving skeletal, ocular, neurological, respiratory, oro-dental, cardio- vascular system but preserved normal mentation and intelligence with typical delayed clinical manifestations usually in 2nd or early 3rd decade. Type- VI also involves the most extensive multisystem clinical cascade with osteoarticular manifestations like short stature, dysostosis multiplex, joint stiffness or contracture, kyphoscoliosis, genu valgum deformity, hip dysplasia and ocular abnormalities representing as corneal clouding, refractive error problems, optic atrophy, optic disc swelling, retinopathy, glaucoma where neurological manifestations are notably ventriculomegaly or hydrocephalus, myelopathy, carpal tunnel syndrome, spinal cord or nerve root compression, seizures, white matter and perivascular space related abnormalities, and cardiac problems like heart valve insufficiency or stenosis due to cardiac valve thickening, pulmonary hypertension, cardiomyopathy, cardiac conduction system disorders, heart failure, fibroelastosis and respiratory abnormalities most commonly recurrent airway infections, sleep disorders, upper and lower airway obstruction are more predominant. Again, orodental findings like hypoplastic mandible, malposition of unerupted teeth, dentigerous cysts, macroglossia, gingival hyperplasia, high arched palate remains part and parcel of this disease spectrum. Other significant clinical manifestations include hearing loss, otitis media, chronic rhinosinusitis, adenoid hypertrophy, coarse facial features, hepatosplenomegaly, umbilical or inguinal hernia, delayed puberty, hirsutism etc. Among MPS spectrum, type VI is extremely rare next only to type IX variety. Mortality in type - VI MPS is mainly due to respiratory or cardiac complications like fatal respiratory infections and or cardiac valvular abnormalities leading to respiratory or cardiac failure or fatal arrhythmias. The distribution of variant types in the ARSB gene are referred to in Figure 6. The incidence also varies among different populations in different geographical areas. Multisystem clinical involvement like skeletal abnormalities (dysostosis multiplex), corneal clouding and cardiac valve thickening remain the most common clinical features. Interesting fact is that in MPS type - VI mental retardation, intellectual disability and seizure like neurological manifestation is almost absent which is a hallmark.
- Harmatz P, Shediac R (2017) Mucopolysaccharidosis VI: pathophysiology, diagnosis and treatment. Front Biosci (Landmark Ed), 22: 385-406.
- D'Avanzo F, Zanetti A, De Filippis C, Tomanin R (2021) Mucopolysaccharidosis Type VI, an Updated Overview of the Disease. Int J Mol Sci, 22: 13456.
- Neufeld EF, Muenzer J (2001) The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 3421-52.
- Muenzer J (2011) Overview of the mucopolysaccharidoses. Rheumatology (Oxford), 5:v4-12.
- Bradford TM, Litjens T, Parkinson EJ, Hopwood JJ, Brooks DA (2002) Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome): a Y210C mutation causes either altered protein handling or altered protein function of N-acetylgalactosamine 4-sulfatase at multiple points in the vacuolar network. Biochemistry, 41: 4962-71.
- D'Avanzo F, Zanetti A, De Filippis C, Tomanin R (2021) Mucopolysaccharidosis Type VI, an Updated Overview of the Disease. Int J Mol Sci, 22: 13456.
FIGURE 1
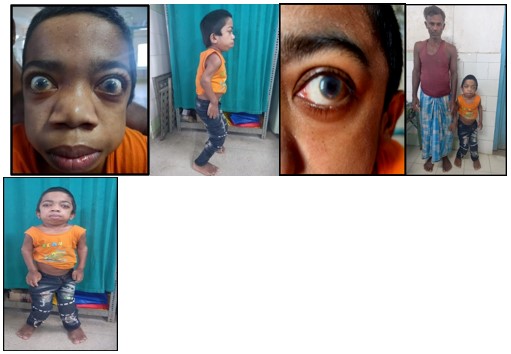
Figure 1: Clinical images of the patient
FIGURE 2
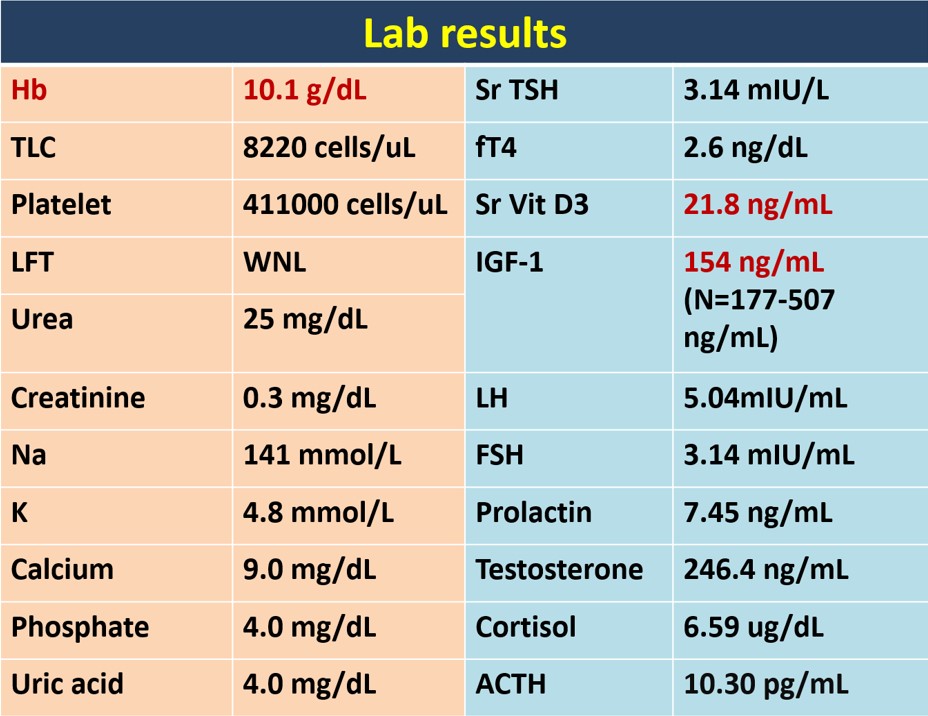
Figure 2: Laboratory investigations
FIGURE 3
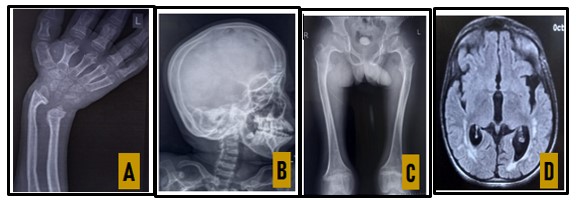
Figure 3: Radiology images
A: X-ray B/L wrist joint: Bone Age = 12-13 yr, suggesting Rickets
B: X-ray Skull (Lat view): Frontal bossing & J shaped sella
C: X-ray of pelvis: Hip dysplasia and femoral epiphyseal widening
D: MRI: Dilated ventricles, Hyperintensities in periventricular, fronto-parietal & temporal white matter.
FIGURE 4
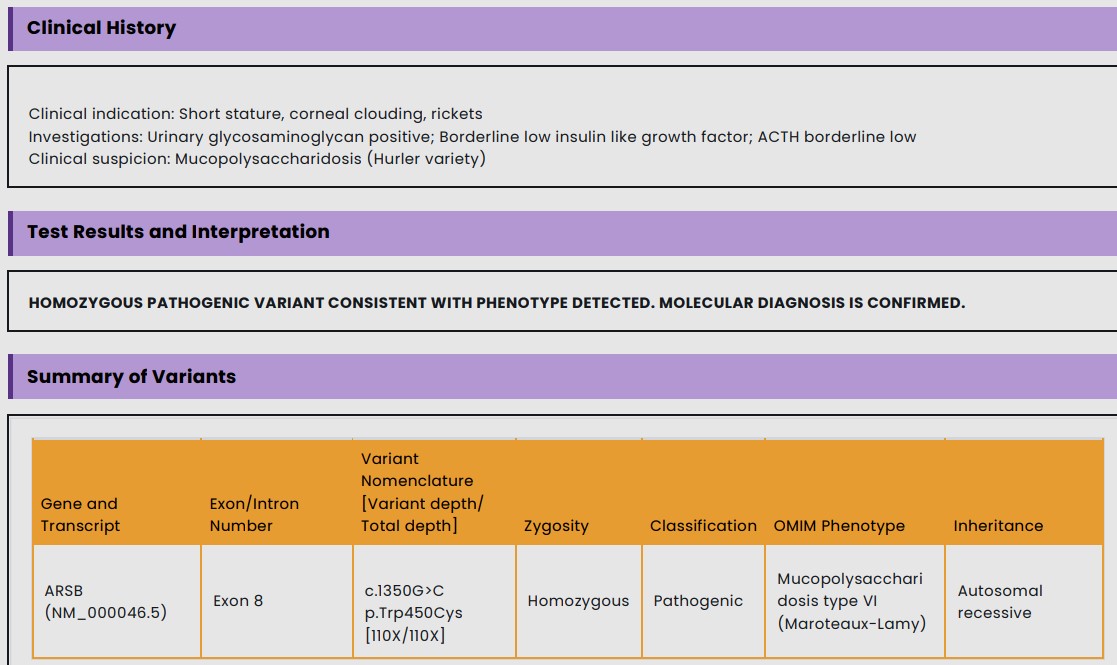
Figure 4: Genetic study
FIGURE 5
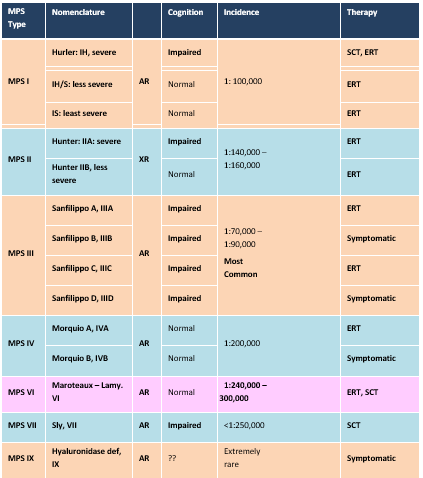
Figure 5
FIGURE 6
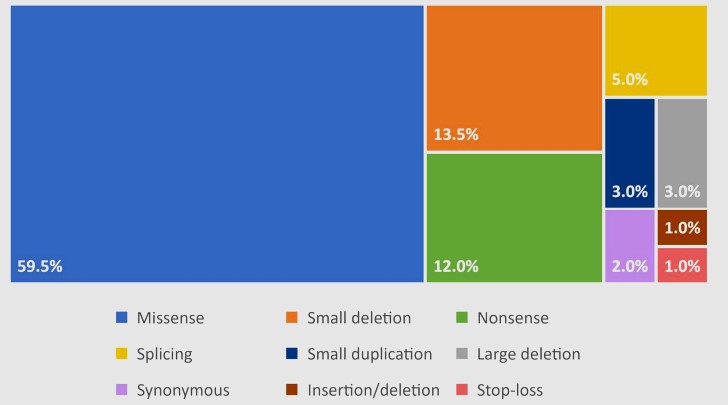
Figure 6: Distribution of Variant Types in the ARSB Gene.
Figures at a glance