Therapy with Growth Hormone in a Patient with Short Stature Secondary to a Mutation in the Acan Gene. About a Case
Received Date: February 09, 2025 Accepted Date: February 24, 2025 Published Date: February 28, 2025
doi: 10.17303/jgdt.2025.1.102
Citation: Carmen Martínez del Río, Olga Pérez Rodríguez (2025) Therapy with Growth Hormone in a Patient with Short Stature Secondary to a Mutation in the Acan Gene. About a Case. J Gen Disor Thera 1: 1-4
Introduction
Aggrecan is a proteoglycan that is part of the extracellular matrix component of the growth plate and cartilage tissue. Pathogenic variants in the ACAN gene have been known for a long time to be responsible for Kimberley-type spondyloepiphyseal dysplasia, aggrecan-type spondylometaphyseal dysplasia, and familial osteochondritis dissecans. However, in 2017 the first pathogenic variants were described in patients with harmonic or disharmonious short stature with mild and variable skeletal defects. No extraskeletal clinical signs seem to be detected.
Case Description
We present the case of an 8-year-old girl undergoing follow-up in pediatric endocrinology consultations of a tertiary hospital in Madrid due to failure to thrive, which was reported more markedly from the age of 3 years.
As a personal history, it was the result of a controlled and normal pregnancy, with consistent control ultrasounds and normal spontaneous vaginal delivery. Small for gestational age anthropometry (Spanish standard deviation score [SDS]: Weight: 2975 g (p29, -0.57 SDS). Length: 46 cm (p2, -2.07 SDS). She did not require resuscitation or admission at birth. She also did not present neonatal jaundice. Normal metabolic tests. Breastfeeding and subsequent introduction of complementary feeding without incident. Up-to-date vaccination calendar and adequate psychomotor development. She has no other medical-surgical history of interest.
Regarding family history, the girl is the only child born to nonconsanguineous parents. Her mother also had short stature that appears secondary to bone dysplasia due to a dysmorphic phenotype (final maternal height after tibial lengthening at 9 years: 140cm (-4.04SDS)). Grandmother mother with final height: 140cm (-4.04 SDS). Paternal grandmother and aunt died of colorectal cancer. (*genealogy chart attached: Figure 1)
In the physical examination of the first visit, a peculiar phenotype with triangular facies, highforehead with frontal bossing and low-set ears was described. Her height and weight at 5 years and 3 months old were 97.1cm (p<1, -3.24 SDS) and 14Kg (p5, -1.71 SDS), respectively. Head circumference: 47.5cm (p<1, -2.45 SDS). Sitting height to standing height ratio at diagnosis 0.561 (p55, 0.15 SDS). Upper to lower segment ratio: 1.1 (normal). Arm Span 99 cm (normal). Arm length 17cm (p<1, -4.48 SDS). Forearm length: 14 cm (p<1, -2.87 SDS). Physical examination revealed a Tanner stage 1.
During the following visits, laboratory serum analysis was performed: complete blood count, biochemistry profile, urinalysis, and thyroid hormone testing were normal. The girl also had negative celiac disease serology. IGF-1 was low in the first measurement, so a GH secretion test was requested, ruling out the deficiency. IGF-1 was normal in all other measurements.
Her bone age at 4 years and 6 months old according to the standard Greulich-Pyle method was 4 years and 2 months old (corresponding bone age). Skeletal survey was also requested, with no pathological findings.
The analysis in the patient reveals a normal female karyotype (46: XX). MLPA of the SHOX gene revealed no abnormalities. Subsequently, a study was carried out using Sanger sequencing of the pathogenic variants most frequently associated with achondroplasia and hypochondroplasia and the FGFR3 gene with normal results. At 5 years of age and given normal results to date and a family history of short stature, a Next-Generation Sequencing panel (SkeletalSeq V10) was requested; heterozygous variant c.5392C>T was detected in exon 12 in ACAN. At that time, the genetic study of the mother was requested, where the same mutation was found in heterozygosis.
The heterozygous variant c.5392C>T; p. (Gin1798*), causes a change of the amino acid cytosine for thymine at position c.5392 of the Aggrecan protein. It is a previously undescribed pathogenic variant that justifies the short stature of our patient.
At 5 years and 3 months with height 97.1cm (p<1, -3.24 SD) it was decided to start treatment with GH (0.03mg/Kg/day) presenting a significant increase in growth rate during the two subsequent years without presenting effects side effects or acceleration of bone age. At the present time (8 years and 1 month) the girl’s height is 116 cm (p<1, -2.47 SD). (*height percentile chart is attached: Figure 2)
Conclusion
Aggrecan and type II collagen are fundamental components of articular cartilage. Alterations in the ACAN gene are, after SHOX haploinsufficiency, the most common underlying monogenic cause of idiopathic short stature. Treatment with GH in this group of patients has shown efficacy similar to that observed in patients diagnosed with idiopathic short stature. In our case, it has been a good Growth Response to Growth Hormone Treatment during the first 2 years, with significant improvement in height and no associated side effects.
- Quintos JB, Guo MH, Dauber A (2015) Idiopathic short stature due to novel heterozygous mutation of the aggrecan gene. J Pediatr Endocrinol Metab 28: 927-32.
- Hu X, Gui B, Su J, Li H, Li N et al. (2017) Chinese Genetic Short Stature Consortium, Li C, Luo J, Fan X, Ding Y, Li J, Wang J, Wang X, Chen S, Shen Y. Novel pathogenic ACAN variants in non-syndromic short stature patients. Clin Chim Acta. 469: 126-9.
- Kim TY, Jang KM, Keum CW, Oh SH, Chung WY (2020) Identification of a heterozygous ACAN mutation in a 15-year-old boy with short stature who presented with advanced bone age: a case report and review of the literature. Ann Pediatr Endocrinol Metab 25: 272-6.
- Mancioppi V, Prodam F, Mellone S, Ricotti R, Giglione E et al. (2021) Retrospective Diagnosis of a Novel ACAN Pathogenic Variant in a Family with Short Stature: A Case Report and Review of the Literature. Front Genet 12: 708864.
- Clinical Characterization of Patients with Autosomal Dominant Short Stature due to Aggrecan Mutations. Gkourogianni A, Andrew M, Tyzinski L, Crocker M, Douglas J et al. (2017) J Clin Endocrinol Metab 102: 460-9.
FIGURE 1
Figure 1: Genealogy chart
FIGURE 2
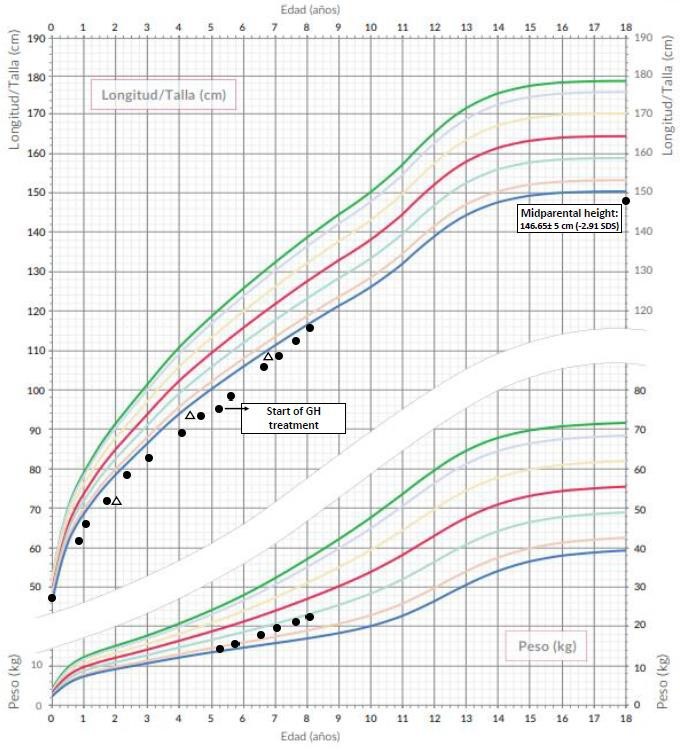
Figure 2: Height percentile chart
Figures at a glance