A Case Report of Fetal Genetic Diagnosis of Sanfilippo Syndrome
Received Date: February 14, 2023 Accepted Date: March 14, 2023 Published Date: March 17, 2023
doi: 10.17303/jgdt.2021.2.101
Citation: Jacqueline A Shah, Santhosh Kumar (2023) A Case Report of Fetal Genetic Diagnosis of Sanfilippo Syndrome. J Gen Disor Thera 2: 1-6
Introduction
Mucopolysaccharidosis type IIIB (MPS III), also known as Sanfilippo syndrome, is a rare autosomal recessive disorder caused by deficiency of Alpha-N-acetylglucosamine required for breakdown of heparan sulfate by lysosomes leading to accumulation of glycosaminoglycans in tissues leading to progressive cellular damage. It that primarily affects the brain and spinal cord, characterized by neurodegeneration, behavioral problems, mild skeletal changesand shortened life span.
Case report
A 4-year-old male child was brought to our OPD with complaints of developmental delay and inability to bear weight on his lower limbs.
The child was the first born child of a second degree consanguinously married couple. Born by normal vaginal delivery with a birth weight of 2Kg and was discharged without any NICU admission. The mother noticed regression of milestones and delayed development of future milestones at 10 months of age. He attained neck holding only at 10 months of age, could sit with support at 19 months of age, could sit without support at 2 years, crawled at 2years 3months and currently is able to stand only with support.Fine motor skills are delayed with only ability to hold objects.Language and social milestones are also delayed. He also has behavioural problems with autistic features, restlessness,hyperkinetic disorder and mental retardation. No visual fixation present.
On examination, the child had microcephaly (head circumference-45cm), short stature, coarse facial features,rocker bottom feet, crowding and overlapping of toes,asymmetry of limbs with shortening of left lower limb, mil hepatomegaly, gibbus deformity of spine. In view of the above findings, MPS was suspected and sent for genetic evaluation.At the time of suspected diagnosis mother was gravida 2 with 16 weeks of gestation, a genetic diagnosis was very important to prevent second child with the same diagnosis.
Genetics
Gene study by whole gene exome sequencing revealed homozygous mutation of NAGLU gene on chromosome 17:40696031 with a prevalence of 1:250000 live births.The missense variant p.N669K in NAGLU(NM_000263.3) is novel which is causes nucleotide C at position 2007 to be changed to nucleotide G. This causes a change in amino acid sequence of the resultant protein from Asparagine to Lysine at position 669. This mutation was confimed by Sanger Sequencing.
The mother spontaneously conceived again and amniocentesis done revealed heterozygous mutation in the NAGLU gene showing a frameshift insertion NM_000263.4 (NAGLU):c.1915_1916delinsTT(p.Glu639Leufs*46) which has not been reported previously as a pathogenic variant or benign variant. The p. Glu639Leufs*46 variant is novel in gnomAD All. This region is critical to protein function and hence classified as likely pathogenic. But anomaly scans revealed normal fetus.
Discussion
Sanfilippo Syndrome also known as Mucopolysaccharidoses III is a rare autosomal recessive disorder caused by mutation responsible for breakdown of heparan sulfate resulting in neurological pathology[1]. There are four subtypes of Sanfilippo syndromes based on the mut a t e d g e n e a n d e n z y m e d e f i c i e n c y . T y p e A (OMIM#252900), type B (OMIM#252920), type C (OMIM#252930), and type D (OMIM#252940). Sanfilippo Syndrome type B is a rare autosomal recessive disorder due to deficiency of alpha-N-acetylgucosaminidase which is required for lysosomal breakdown of heparan sulfate[2] caused by homozygous or compound heterozygous mutation in the NAGLU gene on chromosome 17q21 with an incidence of 1 in 70000 live births[3]. At least 118 mutations in the NAGLU gene have been found to cause mucopolysaccharidosis type IIIB. Heparan sulphate molecules that have not been destroyed build up in lysosomes, causing cellular malfunction and pathology in a number of organs, with severe central nervous system degeneration as the primary phenotypic hallmark.
Affected children have profound mental retardation,intractable behavior problems, destructive and aggressive behavior, hyperactivity, pica, seizures, deafness, loss of vision, and an inability to sleep and eventually dementia [4].Somatic symptoms are mild and death usually occurs in second decade. They also have skeletal abnormalities with osteonecrosis of the femoral head in severe cases [5]. Coarse facial features, thick lips, synophrys and stiff joints are also observed [6]. Type A is usually the most severe subtype, characterized by earliest onset, rapid clinical progression with severe symptoms, and short survival [7]. The median age of death for children afflicted with type A is 15.4 ± 4.1years. [8]
A urinalysis can show elevated levels of heparan sulfate in the urine. The diagnosis may be confirmed by enzyme assay of skin fibroblasts and white blood cells. Prenatal diagnosis is possible by chorionic villus sampling or amniocentesis.People with two working copies of the gene are unaffected. People with one working copy are genetic carriers of Sanfilippo syndrome. They have no symptoms but may pass down the defective gene to their children. People with two defective copies will suffer from Sanfilippo syndrome [9].
Treatment is supportive. If an early diagnosis is made bone marrow replacement may be beneficial. Several newer modalities and gene based therapies are in development [10].
The median age of death of Sanfilippo syndrome type A was 15.22+/- 4.22, Type B 18.91+/-7.33 years, type C 23.43 +/-9.47 years[11].
As the patient ages and the disease advances, providing care for Sanfilippo B patients has an impact on all facets of family life[12]. Since it is a disease with high morbidity and short lifespan the burden on the caregivers is also high with burden of daily care and psychological stress.Hence prenatal evaluation plays a major role in early diagnosis and decision making in such cases and helps in reducing the burden of disease in the community.
- Sanfilippo SJ, Good RA, Podosin R, Langer L (1963) Mental Retardation Associated with Acid Mucopolysacchariduria (Heparitin Sulfate Type) J. Pediatr-Us 63: 837-8.
- Schmidtchen A, Greenberg D, Zhao HG et al. (1998) NAGLU Mutations Underlying Sanfilippo Syndrome Type B.The American Journal of Human Genetics 62: 64-9.
- Tomatsu S, Pitz S, Hampel U (2019) Ophthalmological Findings in Mucopolysaccharidoses. Journal of Clinical Medicine 8: 1467
- Valstar MJ, Bruggenwirth HT, Olmer R, Wevers RA,Verheijen FW et al. (2010) Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype.Journal of Inherited Metabolic Disease 33: 759-67
- Breyer SR, Vettorazzi E, Schmitz L, Gulati A, von Cossel KM et al. (2021) Hip pathologies in mucopolysaccharidosis type III. Journal of Orthopaedic Surgery and Research.
- Cleary MA, Wraith JE (1993) Management of mucopolysaccharidosis type III. Archives of Disease in Childhood 69: 403-6
- Andrade F, Aldámiz-Echevarría L, Llarena M, Couce ML (2015) Sanfilippo syndrome: Overall review. Pediatrics International 57: 331-8
- Tardieu M, Zérah M, Husson B et al. (2014) Intracerebral Administration of Adeno-Associated Viral Vector Serotyperh.10 Carrying Human SGSH and SUMF1 cDNAs in Children with Mucopolysaccharidosis Type IIIA Disease: Results of a Phase I/II Trial. Human Gene Therapy 25: 506-16.
- Meijer OL, Wagemans T, van Lenthe H, Wijburg FA, van Vlies N (2016) Quantity and structure of stored heparan sulfate may affect the nature and course of neuronopathic disease in mucopolysaccharidosis type I and mucopolysaccharidosis type III. Molecular Genetics and Metabolism 117: S79–S80
- Intracerebral Gene Therapy for Sanfilippo Type A Syndrome on clinicaltrials.gov
- Lavery C, Hendriksz CJ, Jones SA (2017) Mortality in patients with Sanfilippo syndrome. Orphanet Journal of Rare Diseases.
- Shapiro Elsa, Lourenço Charles Marques, Mungan Neslihan Onenli, Muschol Nicole, O’Neill Cara et al. (2019) "Analysis of the caregiver burden associated with Sanfilippo syndrome type B: panel recommendations based on qualitative and quantitative data". Orphanet Journal of Rare Diseases 14: 168.
FIGURE 1
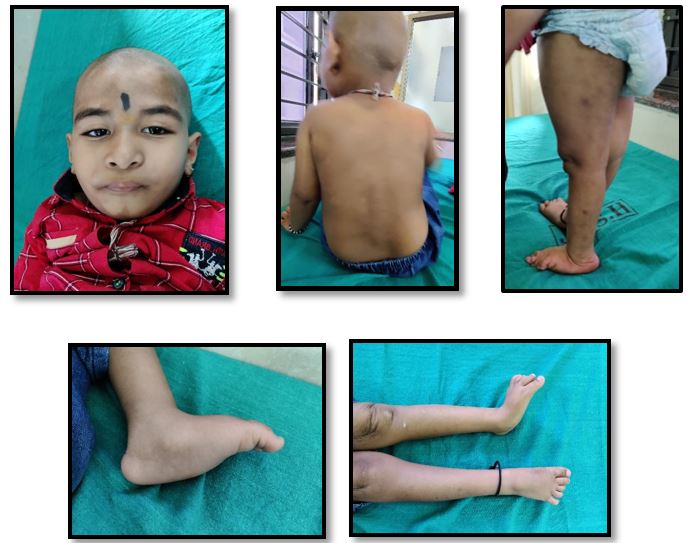
Figure 1:
FIGURE 2

Figure 2:
FIGURE 3
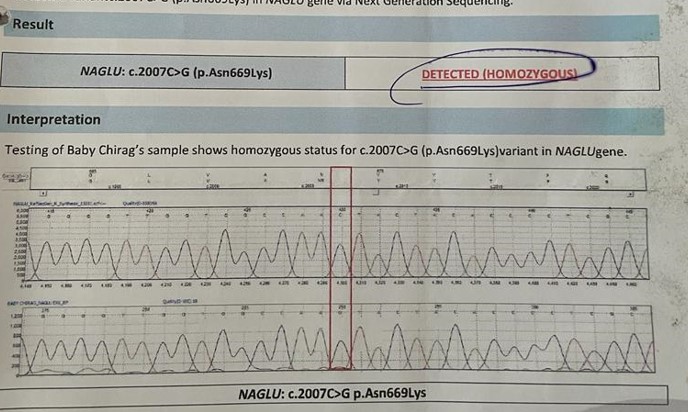
Figure 3:
FIGURE 4

Figure 4:
Figures at a glance