In the Diabetic Rat Heart, Aldosterone and the Autocrine Control of Potassium Currents and Oxidative Stress
Received Date: January 04, 2024 Accepted Date: February 04, 2024 Published Date: February 07, 2024
doi: 10.17303/jmdd.2024.2.102
Citation: Hari Prasad Sonwani (2024) In the Diabetic Rat Heart, Aldosterone and the Autocrine Control of Potassium Currents and Oxidative Stress. J Metab Disord Diabetes 2: 1-14
Abstract
Background and objective: Testosterone has a major impact on heart pathophysiology. The aim of this study was to investigate the effects of cardiac aldosterone on streptozotocin-induced oxidative stress and K+ currents in the diabetic rat heart.
The experiment's methodology: Both transient and sustained K+ currents in ventricular myocytes were measured using a voltage clamp. The amount of aldosterone in cells and plasma was measured using ELISA. Fluorescent dihydroethidium (DHE) was used as a biomarker of oxidative stress to assess superoxide ions.
Significant results: A mineralocorticoid antagonist called spirolactone (1 μM, 5–9 h) significantly shortened the action potential and raised both K+ currents in diabetic males while having no effect on the myocytes of either diabetic female or normal male. Spironolactone's side effects were removed. In female diabetic ovariectomized animals and recovered in those whose bodies had been orchiectomized men. In male diabetic cells, but not in female cells, FAD286 (1 μM, 5–9 h) significantly enhanced K+ currents by inhibiting aldosterone synthase. Spiro lactone and FAD286 significantly reduced oxidative damage in male diabetes cells. Compared to controls, males with diabetes exhibited greater plasma aldosterone levels, whereas females did not. Additionally, there was a little increase in cellular aldosterone. Angiotensin II cellular upregulation was not totally necessary for the rise in aldosterone.
Drawings and conclusions: There was a sex-hormone-dependent, gender-related rise in cardiac cell aldosterone and plasma in male diabetic rats, which resulted in oxidative stress and attenuation of K+ currents. Aldosterone may therefore contribute to cardiac arrhythmias associated with In male diabetic rats, plasma and cardiac cell aldosterone increased in a sexhormone-dependent manner that was related to gender and resulted in oxidative stress and attenuation of K+ currents. Aldosterone may therefore contribute to ventricular arrhythmias associated with diabetes. There was a partial correlation found between higher levels of aldosterone and angiotensin II; nevertheless, residual, angiotensin II-independent aldosterone maintains operating pertinence.
Keywords: Angiotensin II; Pathophysiology; Orchiectomized; Mineralocorticoid
Introduction
Heart function is adversely affected by high aldosterone levels, as demonstrated by numerous basic studies and significant clinical trials [1-3]. Many harmful effects of aldosterone include the production of inflammation, fibrosis, and oxidative stress [4,5].
Furthermore pro-arrhythmic is aldosterone [6]. This is caused by direct impacts on many ionic currents [7] as well as indirect systemic effects including changes in potassium levels or development of fibrosis [6,8].
These modifications are linked to abnormalities in the ECG, including lengthening of recognized risk factors for sudden cardiac death [6,9,10] include the QT interval and increased QT dispersion [6-8]. According to [11], aldosterone antagonists also stop QT prolonging and dispersion as well as the electrical remodeling that occurs in rat hearts after myocardial infarction.
After myocardial infarction, in both systolic and diastolic heart failure [3,12,13], and in hypertension, elevated cardiac aldosterone levels have been found. The Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacious and Survial Study (EPHESUS) and the Randomized Aldactone Evaluation Study (RALES), two sizable investigations, have demonstrated that inhibition of Patients with heart failure had lower mortality and morbidity when they have a mineralocorticoid receptor [13,14]. Still up for debate, though, is where the rise in cardiac aldosterone came from [15,16].
On the one hand, there is proof that the genes responsible for producing aldosterone in cardiac cells are expressed [17,18], regardless of the amount of the hormone in the blood [19-23]. However, some data indicate that pathology-driven higher levels may reflect greater binding or uptake from the plasma, rather than local synthesis, given on the relatively low cardiac levels of aldosterone [2,3,16,24,25].
A portion of the contentious data could stem from variations in the species/strains examined [15,16]; alternatively, it could be because cardiac cells don't normally synthesize aldosterone and only do so in pathological circumstances [15;16,22,26]. Diabetes is a significant pathology where aldosterone may be involved. The prevalence of diabetes is rising quickly [27,28].
This condition can cause major cardiovascular problems, such as the emergence of a unique cardiomyopathy. According to [29], mechanical and electrical damage result in a high diabetes is an independent predictor of death, independently to preexisting hypertension or ischemic heart disease, due to the prevalence of heart failure [30]. Increased plasma aldosterone may be linked to diabetes [29,31].
Aldosterone receptor inhibition has been shown to improve heart function. There is evidence to show that high levels of aldosterone and hyperglycemia amplify each other's harmful effects [5,32]. Attenuation of K+ currents, which are responsible for the repolarization of the ventricular action potential, is one of the electrophysiological alterations associated with diabetes [33,34]. The prolongation of the QT interval in the diabetic human ECG (Ewing) may be partially explained by the ensuing prolongation of the action potential [35,36].
Numerous kinds of diabetes-related arrhythmias have known substrates in the form of QT lengthening and increased QT dispersion [36,37]. In rat and human cardiac myocytes, diabetes has also been demonstrated to increase angiotensin II [28,38,39]. Diabetes may cause an elevated risk of sudden cardiac death due to an increase in angiotensin II [10,40,41]. This could be partly due to an inhibition of K+ currents, as in the type I diabetes rat caused by streptozotocin (STZ) [42] or the diabetic rabbit induced by alloxan [43]. This attenuation could be caused in part by due to increased levels of angiotensin II, which have been demonstrated to directly attenuate several K+ current types [44,45]. Angiotensin II induces oxidative stress, which has an impact on K+ currents [46]. Our previous study revealed a key gender difference: in myocytes from diabetic females, angiotensin II levels are not elevated [42]. As a result, there is less oxidative stress and less attenuation of K+ currents than in diabetic males. Higher levels of angiotensin II can be beneficial because it is a significant regulator of aldosterone synthesis [47], could result in elevated levels of aldosterone. Nevertheless, angiotensin II may not always be necessary for an increase in aldosterone levels [6]. According to recent reports [48], angiotensin II receptor knockout mice may exhibit persistent cardiac aldosterone synthesis. Additionally, heart failure patients may have elevated plasma aldosterone levels despite complete inhibition of angiotensin-converting enzyme (ACE).
Discussion
Overview of the Outcomes
To the best of our knowledge, our findings are the first to demonstrate the role aldosterone plays in regulating cardiac K+ currents and oxidative stress in the context of diabetes. The current work demonstrates that aldosterone-dependent K+ current attenuation and action potential extension occur as part of the diabetic heart pathology, which is consistent with the reported attenuation of K+ currents by exogenous (in vitro) aldosterone [49]. Our findings demonstrate a gender-specific increase in currents that is attained through the inhibition of aldosterone synthase with FAD286 and the inhibition of mineralocorticoid receptors with spironolactone. Additionally, both substances lessen oxidative stress in the ventricular cells of male diabetic rats. An important factor in the gender-dependent impacts appears to be sex hormones. It appears that female sex hormones prevent K+ current regulation by aldosterone suppression, since diabetic male and female ovariectomized cells exhibit comparable responses. In contrast, while spironolactone effects are missing in male orchiectomized diabetics, androgens in males appear to contribute to the present attenuation by aldosterone. Lastly, the data demonstrate that cellular aldosterone is also dramatically decreased by a considerable fall in angiotensin II concentration caused by prolonged ACE inhibition. Nevertheless, spironolactone continues to increase K+ currents, indicating that aldosterone may have some effects that are not dependent on angiotensin II.
Study limitations
Results from rat models must be applied with extreme caution to humans. This study's complement of K+ currents that determine repolarization differs in humans and rats, with the human ventricle having a greater prominence of the slow and fast currents IKr and IKs. Nevertheless, a type I diabetic rabbit model likewise exhibits attenuation of both these currents [43]. Furthermore, there are a number of similarities between the STZ rat model and human diabetes. These include the increase in cardiac angiotensin II levels in rats [38] and humans [39], as well as the prolonging of the QT interval in rat [50] and human [36] ECGs. It's interesting to note that angiotensin II may reduce IKs [44], highlighting potential similarities between renin-angiotensin-aldosterone system activity and electrophysiological abnormalities in humans and rats. The tiny levels of cellular aldosterone and angiotensin II, which cause variation in ELISA quantification, are another research constraint. Variability in the amount of changes in currents and oxidative stress is also caused by further variation in the degree of disruption in these chemicals' levels. However, the combination of techniques, including oxidative stress, ELISA, and current measurements, helps to partially offset this variability. Moreover, the impossibility of full quantification hinders research on the angiotensin II-independent aldosterone action because even with total ACE inhibition, there may be more channels for the production of angiotensin II, which could raise levels of aldosterone.However, our findings indicate that functionally meaningful amounts of aldosterone are still able to influence K+ currents in the diabetic heart even after extremely significant ACE suppression. Lastly, while type II diabetes accounts for the majority of cases of diabetes in humans, our work used a type I diabetes model. However, recognition of the overlap between the two categories is expanding (see, for instance, Severson, 2004). Thus, hyperglycemia, pancreatic β cell secretory abnormalities, oxidative stress, comparable ECG alterations, and angiotensin system activation are shared by both kinds of diabetes.
Meaning and Interpretation
Our findings are consistent with a growing understanding of the detrimental effects of high aldosterone in cardiac pathologies, including diabetes [5] and heart failure [4,1]. Numerous studies that highlight the advantages of mineralocorticoid antagonists-which are now widely used as a therapeutic intervention-also highlight the harmful effects of increased aldosterone [14,51]. These positive outcomes occur regardless of the reduction in hypertension brought about by these substances [51]. Because it might worsen the damage caused by hyperglycemia, an increase in testosterone may be especially dangerous in people with diabetes [5]. On the other hand, hyperglycemia can make the negative effects of aldosterone worse [32,52]. Our findings imply that the known pro-arrhythmic characteristics of diabetic complications may be influenced by the electrophysiological effects of aldosterone, specifically the prolongation of the QT interval in the ECG and the attenuation of outward K+ currents [8,9,11,37,40,49]. Therefore, aldosterone receptor blockage by mineralocorticoid antagonists is advantageous for diabetic patients [5]. According to our research, some of these advantages might be associated with an absence of aldosterone's arrhythmogenic effects. Additional research demonstrates that long-term suppression of the aldosterone receptor impedes the decrease in outflow currents that occurs after myocardial infarction in addition to fixing the ECG's QT anomalies [11].
Cellular Aldosterone and Plasma
While the evidence for extra-adrenal synthesis of aldosterone [17,23,26] is still controversial, the elevation of plasma aldosterone in cardiac pathology appears established [14,31,48,53,54]. Our findings verify that male diabetic rats have higher plasma levels of aldosterone. Male diabetics also exhibited a tendency for cellular aldosterone to rise. Nevertheless, as its source can be increased absorption, an increase in cellular aldosterone does not necessarily signify improved cellular synthesis [25]. Which demonstrate current augmentation with the aldosterone synthase inhibitor FAD286, unmistakably indicates increased cellular production under diabetic circumstances, since isolated single cells are used to measure these effects. The decrease in oxidative stress in isolated cells after exposure to FAD286 independently confirms this. In diabetics, an increase in either plasma or cellular aldosterone would be adequate to reduce potassium currents. According to [49] aldosterone may reduce the transient K+ current in normal rat cells by altering the synthesis of channel protein, which is triggered by variations in calcium levels. These effects don't show up until 48 hours following incubation. It is likely that the increased levels of aldosterone in cardiac cells seen in this study have comparable effects on K+ channels. Consequently, only after more than five hours do Ipeak and Isus increase when aldosterone production or activity is suppressed, suggesting that, similar to quinapril, an ACE inhibitor, fresh channel protein synthesis is necessary [34].
Disparities in Gender
The lack of effect of FAD286 or spironolactone on K+ currents in the cells from these rats is consistent with the fact that neither cellular nor plasma aldosterone increases in female diabetic rats. This finding is in line with our previous research [33,34] which shown variations in the response to ACE inhibition and oxidative stress between genders [35]. It was suggested that estrogen's inhibition of the RAS was the cause of this discrepancy [55]. Our findings imply that oestrogen also suppresses the production of aldosterone, as evidenced by the return of spironolactone-induced augmentation of K+ currents in female diabetic ovariectomized patients. On the other hand, it has been proposed that androgens cause or worsen a number of gender disparities [55,56]. It has been demonstrated that androgens activate certain RAS components [55,57]. Our findings are the first to imply that some gender differences originate from androgen effects on the RAS rather than (or in addition to) direct effects on K+ currents themselves as in [56]. of our study shows that spironolactone has no effect on K+ currents in orchiectomized rats. It is therefore suggested that androgens play a role in the acquired sensitivity of K+ currents to aldosterone suppression in males with diabetes. The creation of diabetes appears to have a lesser (or missing) effect of elevating aldosterone in orchiectomized rats in the absence (or severe reduction) of androgens, so that spironolactone no increases K+ currents for longer.
Aldosterone's Dependency on Angiotensin II
Numerous alterations in heart structure and function are brought about by diabetes [29]. The two main triggers for aldosterone increase are the RAS activation and the rise in angiotensin levels (caused by hyperglycemia). Experiments involving prolonged in vivo ACE inhibition, which results in a significant decrease in aldosterone, provide evidence for this Compared to diabetic animals not receiving quinapril in vivo; the extremely significant reduction in angiotensin II by in vivo quinapril makes K+ currents in cells from diabetic rats insensitive to further in vitro quinapril. Even Nevertheless, after angiotensin II suppression, aldosterone receptor blockage substantially increases both K+ currents It is plausible that minute quantities of leftover angiotensin-II all of the aldosterone's side effects that are still there after quinapril medication. However, in other investigations, including those involving mice that had the angiotensin II receptor 1A removed [26] and complete suppression of ACE, it was discovered that the rise of plasma aldosterone persisted. In the latter instance, aldosterone synthesis may be mediated by additional angiotensin II receptors or ACE-independent angiotensin II synthesis. Studies demonstrating additive ameliorative effects of angiotensin II and aldosterone inhibition offer more evidence in favor of angiotensin II-independent aldosterone action [51]. Aldosterone production that is angiotensin II-independent may be caused by increased sympathetic activity [58] or raised endothelin-1 levels. Another autocrine modulator that we previously demonstrated to be involved in the modulation of K+ current is endothelin-1 [34]. Our unpublished results indicate that endothelin-1 was elevated in the cells of STZ diabetic rats, and this may be one reason for the elevated aldosterone levels. Our findings, which demonstrate that aldosterone affects K+ currents in the presence of diabetes without the angiotensin II impact, are significant because they shed light on the intricate regulation of K+ currents. Therefore, there are several ways to repair anomalies in K+ current and potentially QT [11]. The investigation of the connections between variations in the levels of various autocrine modulators, which can happen without alterations in plasma concentrations [17], which could have significant clinical ramifications. ACE inhibitors are among the frequently used medications that have negative side effects associated with them [59,60]. Therefore, a logical design of therapeutic options for correcting or preventing diabetic problems will be made possible by knowledge of the role and interrelationship of autocrine modulators.
In Summary
Our study shows that elevated cardiac and plasma aldosterone levels cause dysfunction in the diabetic male rat heart. Both arrhythmogenesis and mechanical impairment may be influenced by the way that aldosterone lengthens the action potential and decreases K+ currents. While androgens amplify these effects, estrogen lessens them. Thus, the decrease in aldosterone's localized cardiac effects may account for at least some of the benefits of aldosterone blocking in diabetic patients. Through effects on repolarizing currents, an improvement in repolarizing currents and action potential shortening may lower the frequency of cardiac arrhythmias and indirectly boost contractile activity.
Conflict of Interest
The authors state no conflict of interest.
- Funder JW (2004) Is aldosterone bad for the heart? Trends Endocrinol Metab, 15: 139-42.
- Fiebeler A, Nussberger J, Shagdarsuren E, Rong S, Hilfenhaus G et al. (2005) Aldosterone synthase inhibitorameliorates angiotensin II-induced organ damage. Circulation, 111: 3087-94.
- Ohtani T, Ohta M, Yamamoto K, Mano T, Sakata Y, Nishio M et al. (2007) Elevated cardiac tissue level of aldosterone and miner-alocorticoid receptor in diastolic heart failure: beneficial effects ofmineralocorticoid receptor blocker. Am J Physiol, 292: R946-54.
- Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT (2002) Aldosterone-induced inflammation in the rat heart: role ofoxidative stress. Am J Pathol, 161: 1773-81.
- McFarlane SI, Sowers JR (2003) Aldosterone function in diabetesmellitus: effects on cardiovascular and renal disease. J ClinEndocrinol Metab, 88: 516-23.
- Struthers AD (1999) Why does spironolactone improve mortalityover and above an ACE inhibitor in chronic heart failure?Br J Clin Pharmacol, 47: 479-82.
- Ouvrard-Pascaud A, Sainte-Marie Y, Benitah JP, Perrier R, SoukaseumC et al. (2005) Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation, 111: 3025-33.
- Matsumura K, Fujii K, Kansui Y, Arima H, Iida M (2005) Prolongationof the QT interval in primary aldosterone.Clin Exp Pharmacol Physiol, 32: 66-9.
- Ewing DJ, Boland O, Neilson JMM, Cho CG, Clarke BF (1991) Autonomic neuropathy, QT interval lengthening, and unexpecteddeaths in male diabetic patients. Diabetologia, 34: 182-5.
- Rana BS, Lim PO, Naas AA, Ogston SA, Newton RW, et al. (2005) QT interval abnormalities are often present at diagnosis indiabetes and are better predictors of cardiac death than anklebrachial pressure index and autonomic function tests. Heart, 91: 44-50.
- Yee KM, Pringle SD, Struthers AD (2001) Circadian variation in the effects of aldosterone blockade on heart rate variability and QTdispersion in congestive heart failure. J Am Coll Cardiol, 37: 1800-7.
- Mizuno Y, Yoshimura M, Yasue H, Sakamoto T, Ogawa H, et al. (2001) Aldosterone production is activated in failingventricle in humans. Circulation, 103: 72-7.
- Pitt B, Remme WJ, Zannad F, Neaton J, Martinez F, et al. (2003) Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med, 348: 1309-21.
- Struthers AD (2004) Aldosterone in heart failure: pathophysiology and treatment. Curr Heart Fail Rep, 1: 171-5.
- Mizuno MJ, Clyne CD, Cole TJ, Funder JW (2001) Cardiac steroido-genesis in the normal and failing heart. J Clin Endocrinol Metab, 86: 5121-6.
- Gomez–Sanchez EP, Ahmad N, Romero DG, Gomez–Sanchez CE (2004) Origin of aldosterone in the heart. Endocrinol, 145: 4796-802.
- Slight SH, Joseph J, Ganjam VK, Weber KT (1999) Extra-adrenal mineralocorticoids and cardiovascular tissue. J Mol Cell Cardiol, 31: 1175-84.
- Casal AJ, Silvestre JS, Delcayre C, Capponi AM (2003) Expression and modulation of steroidogenic acute regulatory protein messenger ribonucleic acid in rat cardiocytes and after myocardial infarction. Endocrinology, 144: 1861-8.
- Silvestre JS, Heymes C, Oubenaissa A, Robert V, Aupetit-Faisant B et al (1999) Activation of cardiac aldosterone production in rat myocardial infarction. Circulation, 99: 2694-701.
- Silvestre JS, Robert V, Heymes C, Aupetit-Faisant B, Mouas C et al. (1998) Myocardial production of aldosterone andcorticosterone in the rat. Physiological regulation. J Biol Chem, 273: 4883-91.
- Delcayre C, Silvestre JS, Garnier A, Oubenaissa A, Cailmail S, Tatara E, et al.(2000). Cardiac aldosterone production and ventricular remodeling. Kidney Int, 57: 1346-51.
- Mizuno Y, Yasue H, Yoshimura M, Harada E, Fujii H, Nakamura S, et al. (2005) Adrenocorticotrophic hormone is produced in the ven-tricle of patients with essential hypertension. J Hypertens, 23: 411-6.
- Wasywich CA, Webster MW, Richards AM, Stewart RA (2006) Coronary sinus and ascending aortic levels of aldosterone, angiotensin II, and B-type natriuretic peptide in patients with aortic stenosis and in patients with coronary heart disease. Am J Cardiol, 97: 1068–72.
- Chai W, Garrelds IM, de Vries R, Danser AHJ (2006) Cardioprotectiveeffects of eplerenone in the rat heart. Hypertension, 47: 665–70
- Emmett M (2006) Cardiac aldosterone synthesis? Am J Cardiol, 97: 1107–8.
- Katada J, Meguro T, Saito H, Ohashi A, Anzai T et al. (2005) Persistent cardiac aldosterone synthesis in angiotensin II type 1Areceptor-knockout mice after myocardial infarction. Circulation, 111: 2157–64.
- Zimmet P, Alberti GK, Shaw J (2001) Global and societal implications of the diabetes epidemic.Nature, 414: 782–7.
- Lim HS, MacFayden RJ, Lip GYH (2004) Diabetes mellitus, the renin–angiotensin–aldosterone system, and the heart. Arch Intern Med, 164: 1737–48.
- Tomlinson C, Gardiner SM, Hebden RA, Bennett T (1992) Functionalconsequences of streptozotocin-induced diabetes mellitus, withparticular reference to the cardiovascular system. Pharmacol Rev, 44: 103–79.
- Poirier P, Despres JP, Bertrand OF (2006) Identifying which patientswith diabetes should be tested for the presence of coronary arterydisease––the importance of baseline electrocardiogram and exercise testing. Can J Cardiol, 22: 9–15.
- Hollenberg NK, Stevanovic R, Agarwal A, Lansang MC, Price DA et al. (2004) Plasma aldosterone concentration in the patient with diabetes mellitus. Kidney Int, 65: 1435–9.
- Jefic D, Mohiuddin N, Alsabbagh R, Fadanelli M, Steigerwalt S (2006) The prevalence of primary aldosteronism in diabetic patients. J Clin Hypertens, 8: 253–6.
- Shimoni Y, Liu XF (2003a) Sex differences in the modulation of Kþcurrents in diabetic rat cardiac myocytes. J Physiol, 550: 401–12.
- Shimoni Y, Liu XF (2003b) Role of PKC in autocrine regulation of ratventricular Kþcurrents by angiotensin and endothelin. Am J Physiol, 284: H1168–81.
- Shimoni Y, Hunt D, Chuang M, Chen KY, Kargacin G et al. (2005) Modulation of potassium currents by angiotensin andoxidative stress in cardiac cells from the diabetic rat. J Physiol, 567: 177–90.
- Rossing P, Breum L, Major-Pederson A, Sato A, Winding H et al. (2001) Prolonged QT cinterval predicts mortality in patients with type 1 diabetes mellitus. Diabet Med 18: 199–205.
- Abo K, Ishida Y, Yoshida R, Hozumi T, Ueno H, Shiotani H et al. (1996) Torsade de Pointes in NIDDM with long QT intervals. Diabetes Care, 9: 1010.
- Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P et al. (2000) Myocyte death in streptozotocin-induced diabetes in ratsis angiotensin II-dependent. Lab Invest, 80: 513–27.
- Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A et al. (2000) Myocardial cell death in human diabetes. Circ Res, 87: 1123–32.
- El-Atat FA, McFarlane SI, Sowers JR, Bigger JT (2004) Sudden cardiac death in patients with diabetes. Curr Diab Rep, 4: 187–193.
- Fischer R, Dechend R, Gapelyuk A, Shagdarsuren E, Gruner K,Gruner A et al. (2007) Angiotensin II-induced suddenarrhythmic death and electrical remodeling. Am J Physiol, 293: H1242–53.
- Shimoni Y, Liu XF (2004) Gender differences in the levels ofangiotensin II and in its action on multiple pathways of Kþcurrent modulation in diabetic rats. Am J Physiol, 287: H311–9.
- Zhang Y, Xiao J, Lin X, Luo X, Wang H, Bai Y et al. (2007) Ionic mechanisms underlying abnormal QT prolongationand the associated arrhythmias in diabetic rabbits: a role of rapid delayed rectifier Kþcurrent. Cell Physiol Biochem, 19: 225–38.
- Daleau P, Turgeon J (1994) Angiotensin II modulates the delayed rectifier potassium current of guinea pig ventricular myocytes. Pflugers Arch, 427: 553–5.
- Yu H, Gao J, Wang H, Wymore R, Steinberg S, McKinnon D et al. (2000) Effects of the renin–angiotensin system on the currentItoin epicardial and endocardial ventricular myocytes from the canine heart. Circ Res 86: 1062–8.
- Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG et al. (1998) Role of NADH/NADPH oxidase-derived H2O2in angio-tensinII-inducedvascularhypertrophy.Hypertension32: 488–95.
- Michel F, Ambroisine ML, Duriez M, Delcayre C, Levy BI, Silvestre JS (2004) Aldosterone enhances ischemia-induced neovasculariza-tion through angiotensin II-dependent pathway. Circulation, 109: 1933–7.
- Jorde UP, Vittorio T, Katz SD, Colombo PC, Latif F et al. (2002) Elevated plasma aldosterone levels despite complete inhibition of the vascular angiotensin-converting enzyme inchronic heart failure. Circulation, 106: 1055.
- Benitah JP, Perrier E, Gomez AM, Vassort G (2001) Effects of aldosterone on transient outward Kþcurrent density in ratventricular myocytes. J Physiol, 537: 151–60.
- D’Amico M, Marfella R, Nappo F, Di Fillipo C, De Angelis L et al. (2001) High glucose induces ventricular instability and increases vasomotor tone in rats. Diabetologia, 44: 464–70.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J (2005) Additive amelioration of left ventricular remodeling and mole-cular alterations by combined aldosterone and angiotensinreceptor blockade after myocardial infarction. Cardiovasc Res, 67: 97–105.
- Sato A, Funder JW (1996) High glucose stimulates aldosterone-induced hypertrophy via type I mineralocorticoid receptors inneonatal rat cardiomyocytes. Endocrinology, 137: 4145–53.
- Nakayama T, Izumi Y, Soma M, Kanmatsue K (1998) Adrenal renin–angiotensin–aldosterone system in streptozotocin-diabetic rats. Horm Metab Res, 30: 12–5.
- Ustundag B, Cay M, Naziroglu M, Dilsiz N, Crabbe MJC et al. (1999) The study of renin–angiotensin–aldosterone in experi-mental diabetes mellitus. Cell Biochem Funct, 17: 193–8.
- Fischer M, Baessler A, Schunkert H (2002) Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res, 53: 672–7.
- Brouillette J, Trepanier-Boulay V, Fiset C (2003) Effect of androgen deficiency on mouse ventricular repolarization. J Physiol, 546: 403–13.
- Reckelhoff JF, Yanes LL, Iliescu R, Fortepiani LA, Granger JP (2005) Testosterone supplementation in aging men and women: possible impact on cardiovascular–renal disease. Am J Physiol, 289: F941–8.
- Bos R, Mougenot N, Findji L, Mediani O, Vanhoutte PM, et al. (2005) Inhibition of catecholamine-induced cardiac fibrosis by an aldosterone antagonist. J Cardiovasc Pharmacol, 45: 8–13.
- Mann JF, Yi QL, Sleight P, Dagenais GR, Gerstein HC et al. (2005) Serum potassium, cardiovascular risk, and effects of an ACE inhibitor: results of the HOPE study. Clin Nephrol, 63: 181–7.
- Nikpoor D, Duan QL, Rouleau GA (2005) Acute adverse reactionsassociated with angiotensin-converting-enzyme inhibitors; genet-ic factors and therapeutic implications. Expert Opin Pharmacother, 6: 1851–6.
- Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, et al. (2002) Mechanisms of increased vascular superoxide production in human diabetes mellitus. Circulation, 105: 1656–62.
- Menard J, Gonzalez MF, Guyene TT, Bissery A (2006) Investigations ofaldosterone-synthase inhibition in rats. J Hypertens, 24: 1147–55.
- Nerbonne JM (2000) Molecular basis of functional voltage-gated Kþchannel diversity in the mammalian myocardium. J Physiol, 525: 285–98.
FIGURE 1
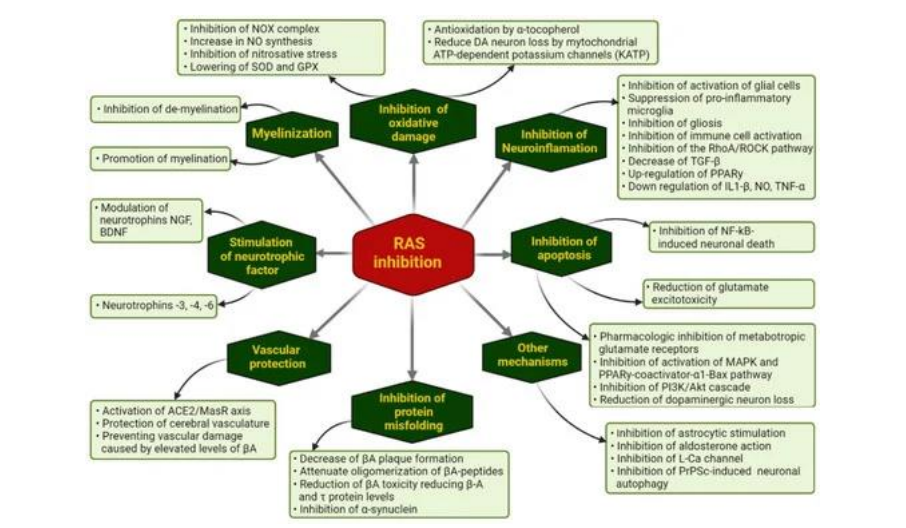
Flow chart: Ways through which pharmacological RAS inhibition may modulate the evolution of NDG diseases
FIGURE 2
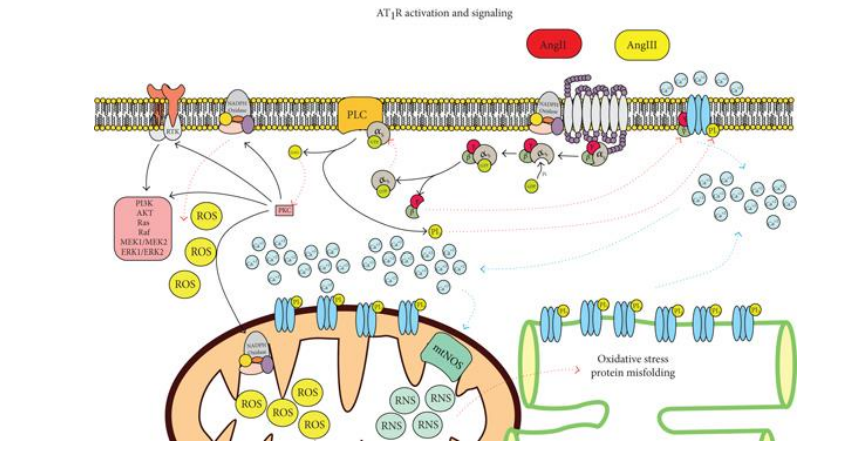
Figure 1: AT1R signaling. The activity of the receptor will stimulate oxidative stress and activation of other signaling pathways
FIGURE 3
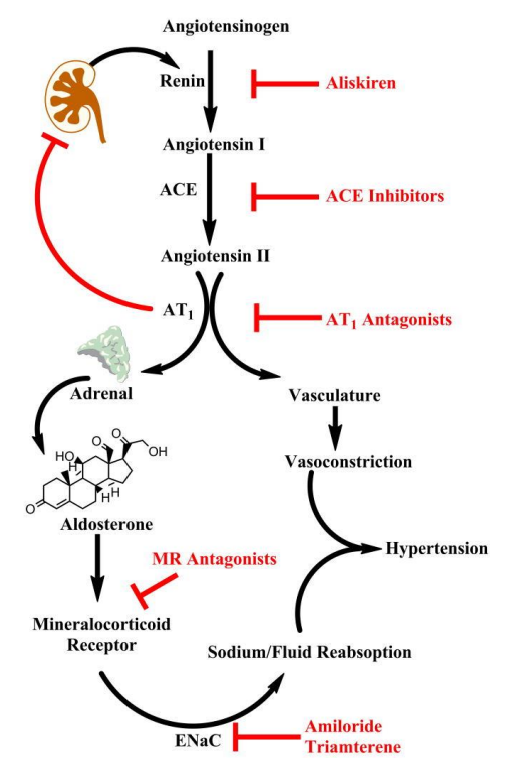
Figure 2: The cascade is initiated by renin secretion, and subsequent conversion of angiotensinogen to angiotensin I (Ang I). Angiotensin II (Ang II) produces vasoconstriction and stimulates adrenal aldosterone secretion via AT1 receptors. Pharmacologic agents are available to block this pathway at nearly every step in the pathway (highlighted in red). Aliskiren is the only currently available renin inhibitor. Angiotensin I converting enzyme (ACE) inhibitors prevent conversion of Ang I to Ang II. Mineralocorticoid receptor (MR) antagonists (e.g. spironolactone, eplerenone) block the effects of aldosterone in the kidney. Downstream targets of aldosterone include the epithelial sodium channel (ENaC) which is upregulated by the MR and mediates sodium reabsorption in the distal kidney
FIGURE 4
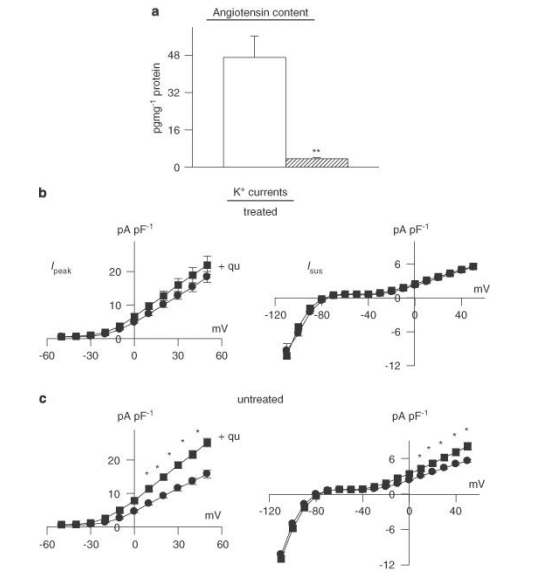
Figure 3: Effects of angiotensin-converting enzyme (ACE) inhibition in vivo. Male rats were given quinapril (6 mg L-1) in their drinking water for 3 weeks prior to induction of diabetes with STZ. Cells were isolated after a further 8-13 days (quinapril continued). (a) Content of angiotensin II was very significantly (P< 0.005) reduced (n=4) in comparison to untreated diabetic males (n=4). (b) The functional implications of this, indicating that in vitro exposure to quinapril (+qu) of cells isolated from rats treated with quinapril in vivo (n=36) had no effect on Ipeak (left) or Isus densities (right), as a function of membrane potential (n=30). This is in marked contrast to the in vitro effect of quinapril in untreated diabetic rats, as shown in (c). In untreated diabetic rats, both Ipeak and Isus are significantly (P< 0.05) augmented by quinapril (*P< 0.05; **P< 0.001).
FIGURE 5
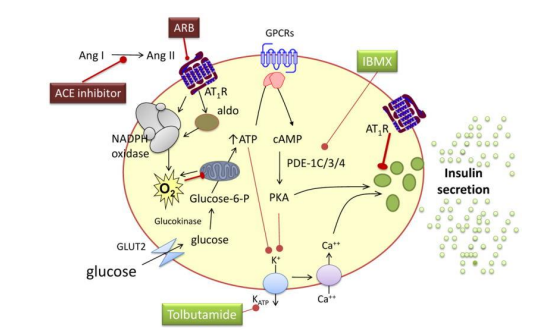
Figure 4: Chronic hyperglycemia and free fatty acids increase the expression of the angiotensin II subtype 1 receptor (AT1R) and uncoupling protein 2, as well as the activity of protein kinase C and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase, leading to inflammation, the formation of reactive oxygen species (O2-), β-cell apoptosis and decreased insulin formation and secretion. These effects are reversed by AT1 receptor blockade (ARB). Aldosterone decreases insulin secretion without affecting the insulin content of β-cells, through a mechanism that also involves reactive oxygen species. This effect is not mediated via the mineralocorticoid receptor. ATP indicates adenosine triphosphate, cAMP cyclic adenosine monophosphate, GLU2 glucose transporter 2, IBMX 3-isobutyl-1-methylxanthine, PDE phosphodiesterase, PKA protein kinase A, GPCR G-protein coupled receptors
FIGURE 6
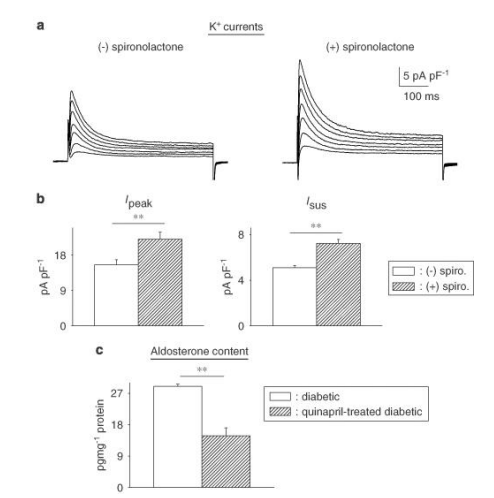
Figure 5: Effects of chronic angiotensin-converting enzyme (ACE) inhibition on aldosterone. (a) Sample current traces (same protocol as above) in cells from diabetic males treated with quinapril in vivo, in the absence of (left), or following (right) spironolactone (1 μM, 6.5 h). (b) Summary data for current densities at +50 mV. Ipeak is shown on the left and Isus on the right. Open bars show mean data from untreated cells (n=31) and hatched bars represent data from cells treated with spironolactone (n=17). Aldosterone inhibition can still significantly augment both currents even when angiotensin II levels are drastically reduced. (c) Aldosterone content (measured by ELISA) is significantly reduced in quinapril-treated diabetic rats (n=4, hatched bars), relative to untreated diabetic rats (n=4, open bars), illustrating that some of the elevation in aldosterone is related to augmented angiotensin II levels. However, there is a functionally significant elevation of aldosterone even in the absence of angiotensin II (**P<0.01).
FIGURE 7
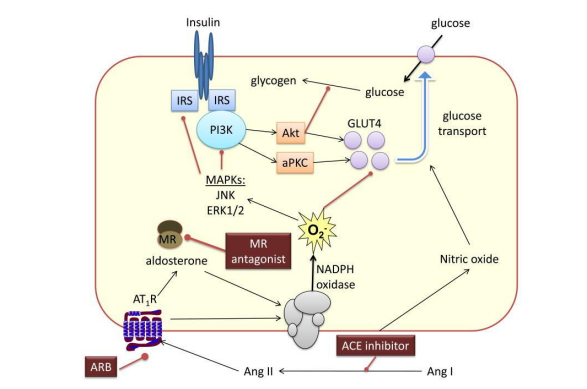
Figure 6: Angiotensin II (Ang II) and aldosterone activate nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase to generate reactive oxygen species (O2-). Activation of redox-sensitive serine kinases such as c-jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK)-1 leads to the phosphorylation of serines in insulin receptor substrate-1 (IRS-1) and decreased interaction with phosphatidylinositol 3-kinase (PI3K). This in turn leads to decreased activation of protein kinase B (Akt) and PKC, decreased translocation of GLUT4 to the membrane and decreased glucose transport. Blockade of the Ang II subtype 1 receptor (AT1R) or the mineralocorticoid receptor prevents these effects of Ang II and aldosterone, respectively. Direct renin inhibitors and angiotensin-converting enzyme (ACE) inhibitors improve insulin resistance by decreasing the formation of Ang II and aldosterone. ACE inhibitors also increase glucose transport via a nitric oxide-dependent mechanism.
Figures at a glance