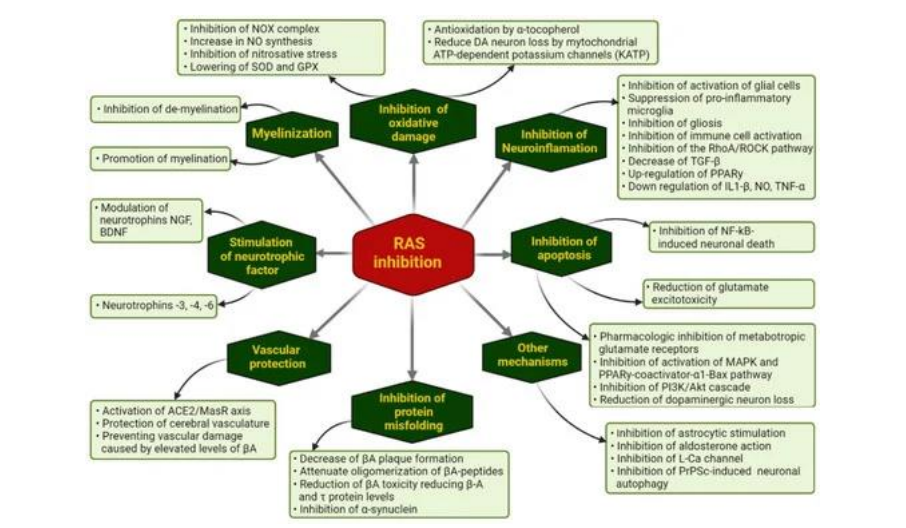
Flow chart: Ways through which pharmacological RAS inhibition may modulate the evolution of NDG diseases
Flow chart: Ways through which pharmacological RAS inhibition may modulate the evolution of NDG diseases
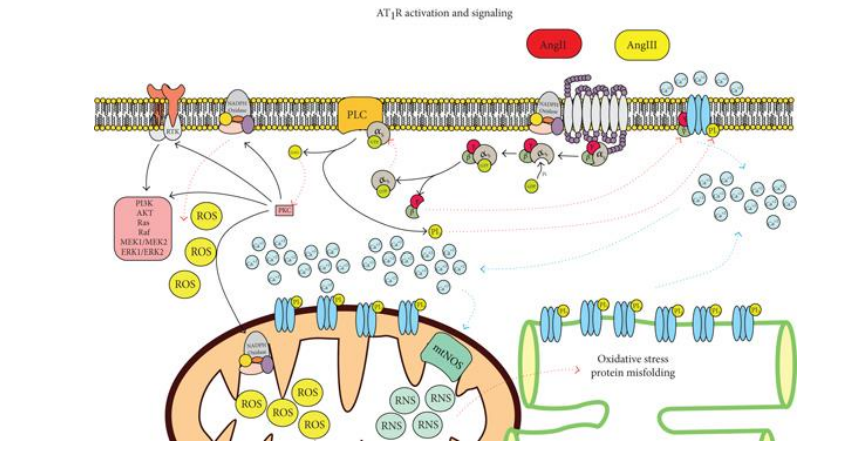
Figure 1: AT1R signaling. The activity of the receptor will stimulate oxidative stress and activation of other signaling pathways
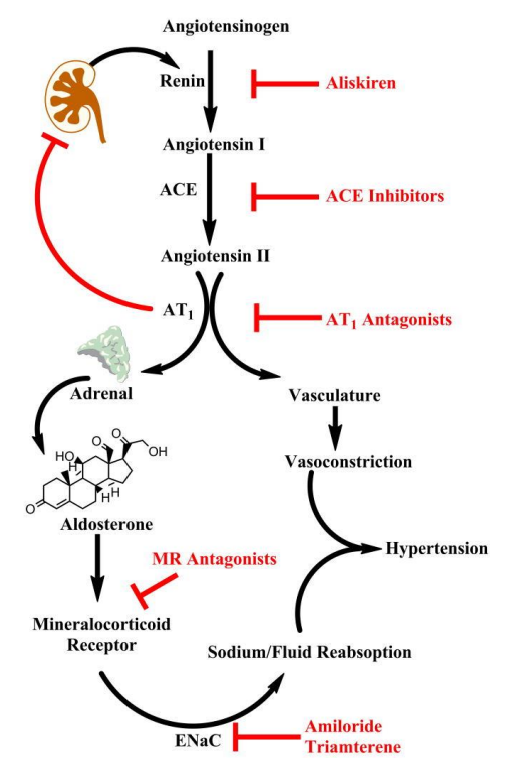
Figure 2: The cascade is initiated by renin secretion, and subsequent conversion of angiotensinogen to angiotensin I (Ang I). Angiotensin II (Ang II) produces vasoconstriction and stimulates adrenal aldosterone secretion via AT1 receptors. Pharmacologic agents are available to block this pathway at nearly every step in the pathway (highlighted in red). Aliskiren is the only currently available renin inhibitor. Angiotensin I converting enzyme (ACE) inhibitors prevent conversion of Ang I to Ang II. Mineralocorticoid receptor (MR) antagonists (e.g. spironolactone, eplerenone) block the effects of aldosterone in the kidney. Downstream targets of aldosterone include the epithelial sodium channel (ENaC) which is upregulated by the MR and mediates sodium reabsorption in the distal kidney
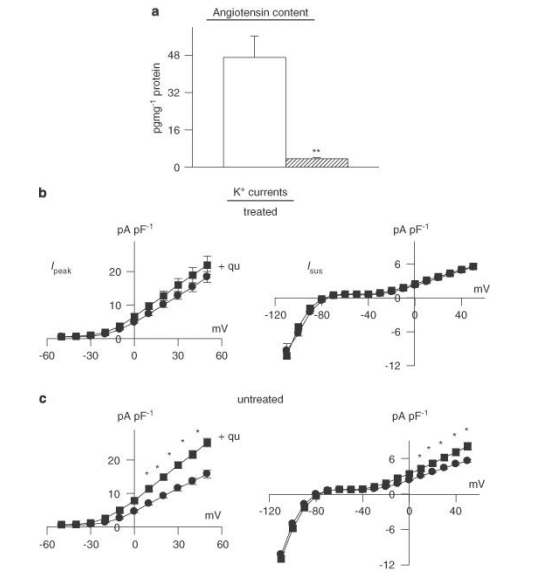
Figure 3: Effects of angiotensin-converting enzyme (ACE) inhibition in vivo. Male rats were given quinapril (6 mg L-1) in their drinking water for 3 weeks prior to induction of diabetes with STZ. Cells were isolated after a further 8-13 days (quinapril continued). (a) Content of angiotensin II was very significantly (P< 0.005) reduced (n=4) in comparison to untreated diabetic males (n=4). (b) The functional implications of this, indicating that in vitro exposure to quinapril (+qu) of cells isolated from rats treated with quinapril in vivo (n=36) had no effect on Ipeak (left) or Isus densities (right), as a function of membrane potential (n=30). This is in marked contrast to the in vitro effect of quinapril in untreated diabetic rats, as shown in (c). In untreated diabetic rats, both Ipeak and Isus are significantly (P< 0.05) augmented by quinapril (*P< 0.05; **P< 0.001).
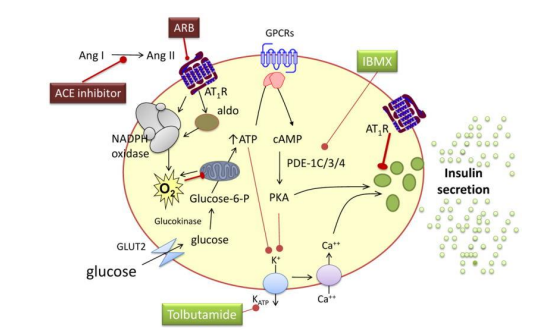
Figure 4: Chronic hyperglycemia and free fatty acids increase the expression of the angiotensin II subtype 1 receptor (AT1R) and uncoupling protein 2, as well as the activity of protein kinase C and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase, leading to inflammation, the formation of reactive oxygen species (O2-), β-cell apoptosis and decreased insulin formation and secretion. These effects are reversed by AT1 receptor blockade (ARB). Aldosterone decreases insulin secretion without affecting the insulin content of β-cells, through a mechanism that also involves reactive oxygen species. This effect is not mediated via the mineralocorticoid receptor. ATP indicates adenosine triphosphate, cAMP cyclic adenosine monophosphate, GLU2 glucose transporter 2, IBMX 3-isobutyl-1-methylxanthine, PDE phosphodiesterase, PKA protein kinase A, GPCR G-protein coupled receptors
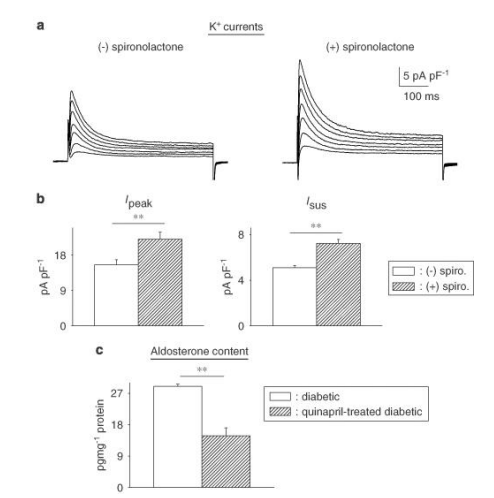
Figure 5: Effects of chronic angiotensin-converting enzyme (ACE) inhibition on aldosterone. (a) Sample current traces (same protocol as above) in cells from diabetic males treated with quinapril in vivo, in the absence of (left), or following (right) spironolactone (1 μM, 6.5 h). (b) Summary data for current densities at +50 mV. Ipeak is shown on the left and Isus on the right. Open bars show mean data from untreated cells (n=31) and hatched bars represent data from cells treated with spironolactone (n=17). Aldosterone inhibition can still significantly augment both currents even when angiotensin II levels are drastically reduced. (c) Aldosterone content (measured by ELISA) is significantly reduced in quinapril-treated diabetic rats (n=4, hatched bars), relative to untreated diabetic rats (n=4, open bars), illustrating that some of the elevation in aldosterone is related to augmented angiotensin II levels. However, there is a functionally significant elevation of aldosterone even in the absence of angiotensin II (**P<0.01).
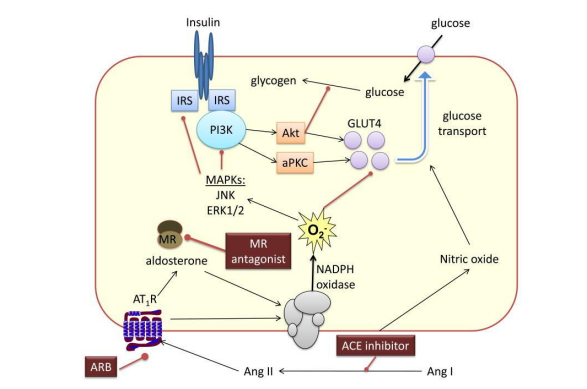
Figure 6: Angiotensin II (Ang II) and aldosterone activate nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase to generate reactive oxygen species (O2-). Activation of redox-sensitive serine kinases such as c-jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK)-1 leads to the phosphorylation of serines in insulin receptor substrate-1 (IRS-1) and decreased interaction with phosphatidylinositol 3-kinase (PI3K). This in turn leads to decreased activation of protein kinase B (Akt) and PKC, decreased translocation of GLUT4 to the membrane and decreased glucose transport. Blockade of the Ang II subtype 1 receptor (AT1R) or the mineralocorticoid receptor prevents these effects of Ang II and aldosterone, respectively. Direct renin inhibitors and angiotensin-converting enzyme (ACE) inhibitors improve insulin resistance by decreasing the formation of Ang II and aldosterone. ACE inhibitors also increase glucose transport via a nitric oxide-dependent mechanism.
Figures at a glance