Additional Arguments against the Correlation Between Blood Glucose Values and Cellular Metabolism
Received Date: February 26, 2024 Accepted Date: March 26, 2024 Published Date: March 29, 2024
doi: 10.17303/jmdd.2024.2.104
Citation: Kornél Simon MD, Attila Cziráki MD (2024) Additional Arguments Against the Correlation Between Blood Glucose Values and Cellular Metabolism. J Metab Disord Diabetes 2: 1-17
Abstract
The persistence of functional and morphological integrity of living organism is supported by the cell metabolism; therefore its diagnostic assessment theoretically is of basic importance. Relationship between blood glucose value and cell metabolism in stress-reaction and diabetic state was analyzed. Authors discuss metabolic characteristics of acute, and repetitive, and chronic stress responses, give definition on terms: eumetabolism, dysmetabolism, hyper metabolism with vs. without balanced energetic equilibrium. In all chronic stress states persistence of stressor triggering the stress-reaction is emphasized as the pathogenetic factor. Acute and repetitive stress are referred to as a health-saving adaptive mechanism, but chronic stress with energetic imbalance in cell metabolism is proposed a health-threatening, progressive, maladaptive phenomenon. Glucose toxicity is suggested the main pathogenetic contributor in eliciting and supporting chronic stress-reaction. The interrelations between metabolic settings of diabetes, and ischemia, and chronic stress, furthermore metabolic effect of acute and chronic hyperglycemia (diabetes) on cell metabolism are disputed. Authors emphasize, that blood glucose value is of crucial importance in governing therapy of the diabetes, but not in assessment of the cellular metabolic state. The relation between the trigger-specific somatic adaptation and the stressor-induced non-specific metabolic adaptation is discussed. Authors suggest, that metabolic characterization of acute, and repetitive, and chronic stress states is a more reliable approach in assessment of cell metabolism (referred to as “metabolic diagnosis”) than blood glucose values associated only with blood glucose transport. Similarities of metabolic settings in starvation, pregnancy, and SGLT2 inhibitor-treatment are presented as an example for clinical application of “metabolic diagnosis”. Chronic stress with preserved energetic balance in cell metabolism, associated with glycogenic mild ketosis, shows beneficial cellular metabolic effect.
Keywords: Stress-Reaction; Diabetes; Eumetabolism; Dysmetabolism; Metabolic Remodeling; Somatic Vs. Metabolic Adaptation
Background
Relationship between the cell metabolism and the blood glucose value portrays an accepted thesis in both physiological (stress-response) and pathophysiological setting (diabetes). Guidelines referencing the most common metabolic disease diabetes, declare unequivocally near-normoglycemia as the main therapeutic target in treatment of diabetes. This thesis suggests that euglycemia (i.e. the normal blood glucose value) is associated with normal metabolic state (eumetabolism), and hyperglycemia is representative of the abnormal metabolism (dysmetabolism). The premise of direct association between blood glucose value and cell metabolism is highly questionable [1]. This review aims to reveal additional insight into relation between blood glucose value and cell metabolism, furthermore to propose a more appropriate approach than blood glucose value for the assessment of cell metabolism.
Central Role of Metabolism in the Living Organism
Metabolism serves as a core function in the human body. Several factors explain this claim. First, the so-- called “serving organs” (cardiovascular, respiratory system and tissue respiration) are responsible for supplying cells with oxygen and nutrients, i.e., for serving metabolism, secondly, the chemical energy (macro erg phosphates) produced by cellular metabolism ensures the function and morphological integrity in all human organs. Metabolism is considered the central physiological process of the living organism: therefore existence of metabolism can be referred to as the "marker of life" (Figure 1). Compared with the wide diagnostic spectrum of "serving and executive organs", the number of crucial methods for studying metabolism, is paradoxically, surprisingly small.
A further argument for the primary role of metabolism premises a common cause of any pathological condition (diseased state) can be defined as the impaired metabolic performance of any origin. The difference between the different pathological entities is, from one perspective, the causes (physical, chemical and/or biological) of impaired metabolic performance are indeed considerably diverse and, secondly, the metabolic damage affects different organs.
The term “cell metabolism” refers to all the biochemical events involved in energy-providing processes of the cell. In our terminology, “eumetabolism” defines a normal metabolic state in which the energy balance of the cell is positive, the resting creatine phosphate/adenosine triphosphate ratio is normal, and the magnitude of the inducible reserve metabolic capacity is also normal. The term “dys-metabolism” defines a pathologic metabolic state in which the energy balance of the cell is negative, the creatine phosphate/adenosine triphosphate ratio is subnormal, and the magnitude of the inducible reserve metabolic capacity is diminished [1].
Stress-Response as a Model for Study the Relationship Between Blood Glucose Value and Cell Metabolism
Selye described stress in 1937 as a complex, nonspecific defense mechanism of the body [2]. During stress adaptation, the organism attempts to remove the stressor i.e. the factor triggering stress, or in the very least, facilitates adaptation to the stressor. The aim is to maintain the health and to ensure survival of the body.
It is well documented that stress activation can be triggered not only by exogenous (physical, chemical and/or biological) causes and endogenous (a chronic disease of vital organs) factors but also by persistent psych mental discomfort (frustration due to civilization stress) [1,3-6].
It is well documented that during stress-reaction in the so-called “serving organs” (cardiovascular, respiratory system), and in the “executive organs” (striated muscular and the central nervous system) the functional performance is enhanced, furthermore characteristic ultrastructure, macro morphological alterations are developed. Additionally there are also well-known changes in inflammatory, hemostaseological and immune systems. Energetic prerequisite for these adaptive mechanisms is ensured by the enhanced metabolic performance of cell metabolism.
Different mechanisms of metabolic adaptation are involved in stress responses induced by the acute presence (acute stress), or by repeated occurrence (repetitive stress), or by permanent persistence (chronic stress) of the stressor.
Study of different stress responses gives opportunity for defining additional information on relation between blood glucose value and cell metabolism.
Relation Between Blood Glucose Values and Cell Metabolism in Acute Stress-Condition
A schematic description of metabolic events in acute stress is summarized below
The activation of stress hormones (catecholamine’s, glucagon, glucocorticoids, etc.,) leads to the mobilization of glucose, free fatty acids and amino acids in order to supply the cellular metabolism. Tissue uptake of glucose and free fatty acids is increased in an obligatory manner, in which blood glucose and free fatty acid concentrations increase to varying degrees dependent upon the severity of the stress involved [3,4,7,8].
Cellular hyper metabolism is characterized by increased glucose and free fatty acid metabolism: cytosolic anaerobic glycolysis activity increases with an enhanced mitochondrial supply of acetyl coenzyme A molecules from the decarboxylation of pyruvic acid as a cytosolic end product.
The carnitine acyl-transferase-mediated mitochondrial fatty acid uptake increases and the quantity of acetyl coenzyme A produced by beta-oxidation rises. Acetyl Co-enzyme A molecules both of free fatty acid and glucose origin are incorporated into the Krebs cycle, in which ATP is produced as a result of terminal oxidation (an electron transport system by a complex redox chain provided by the mitochondrial enzyme system) and aerobic oxidative phosphorylation (saturation of the external electron orbit in a nascent oxygen atom) [4,9-11].
All these processes are carried out in pristine harmony, with maximum efficiency and minimum calcium-ion and oxygen free radical load. This well-coordinated, perfected process is identified as allostatic load (allostasis: equilibrium state): harmoniously coordinated biochemical processes at maximum efficiency can be described by the analogy of an "orchestra conducted by a conductor" [3,4,9,12,13].
The process is apparently a catabolic event due to the depletion of glucose and fat stores, resulting in a decrease in the body's chemical free energy. Cellular metabolism in the allostatic load state is energetically balanced, i.e., it does not create energetic debt. This is the compensated phase of the stress response, in which energy expenditure is counter balanced through metabolic hyperactivity
However, the prolongation of the acute stress activation reaches a point in which the allostatic load state turns into a commonly referred to allostatic overload state. This can be characterized by the fact in which the energetic balance is upset, and the equilibrium of the process becomes negative: macroergic phosphate production cannot counter act its use, and the stress response evolves into a metabolically decompensated state [3,4,8,9,13-15].
The trigger factors behind the energy imbalance can be due to the following
The rate-limiting factor for maximum metabolic performance is oxygen supply. Once this is triggered, several types of "escape mechanisms" are activated.
Instead of consuming free fatty acids, glucose combustion takes precedence. The "rationale" behind this implies the end products of the metabolism of both molecules (glucose and free fatty acid) are carbon dioxide and water. Since the glucose molecule contains more oxygen than the free fatty acid, less exogenous oxygen is needed for complete combustion. This results in a higher ATP per exogenous oxygen ratio for glucose combustion than for fatty acid use [3,12,15].
Acidification due to oxygen deprivation activates the Bohr Effect, which improves oxygen access by shifting the oxygen hemoglobin dissociation curve to the right.
Another trigger for switching to an allostatic overload state may be glucose shortage. The explanation for this is the occurrence of the so-called anaplerosis inhibition [3,10]. Indeed, the oxalacetic acid, which comes about from condensation of the pyruvic acid produced from glucose during anaerobic glycolysis and the carbon dioxide is the sine qua non compound of the Krebs cycle. Insufficient amount of oxalacetic acid deteriorates effectiveness of mitochondrial function. The existence of anaplerosis explains the validity regarding the textbook premise, "fats are combusted in the fire of carbohydrates".
The third trigger is the extreme free fatty acid production due to stress hyperactivity, resulting in toxic fatty acid products which inhibits glucose uptake and commonly-referred to glucose-associated metabolic processes, lead ing to a fall of ATP production (Randle cycle) [3,10,11,16].
In the development of acute stress induced allostatic overload the role of “catecholamine cardiac toxicity” is exemplified by Takotsubo syndrome [17].
Mechanisms involved in improving the efficiency of metabolism are:
The use of hydrogen, due to the increased lactic acid formation from pyruvic acid, promotes NAD regeneration of NADH, which supports the activity of terminal oxidation, hence, macro erg phosphate synthesis [3,12,15,18].
It is highly plausible to suppose, that the efficiency of oxidative phosphorylation is also enhanced by the increased activity of A3 receptors and the commonly identified metabolic sensor: adenosine monophosphate protein kinase [1,3,4,7,8,12,15,19,20]. This assumption is aptly justified by the arguments, as follows: during stress response, the efficiency of the “serving organs” (performance of respiration, stroke volume, heart rate and tissue respiration) is also significantly improved, thus, the rise in metabolic performance reflecting the teleological reason of the whole operation can also be assumed.
Nonetheless, in a state of well-known allostatic overload, the efficiency of mitochondrial function deteriorates, the accumulation of acetyl-Co-A products leads to a tendency to ketosis, the accumulation of pyruvic acid leads to a tendency to lactic acidosis, and the toxic fatty acid products result in a state of lipotoxicity. In the disrupted cellular metabolism the macroergic phosphate availability is restricted to the creatine phosphate reserve and to the ATP production from cytosolic anaerobic glycolysis. In this state, calcium-ion and oxygen free radical loads are increased, leading to a circulus vitiosus-like self-generating increase in dysfunction and ultra-structural damage [1,3,4,8,9,21].
The state of metabolic collapse inevitably parallels with the functional decline.
It should be emphasized, while in the allostatic load state, high blood glucose and fatty acid values coincide with high cellular metabolic performance, in contrast the same parameters in the allostatic overload state coincide with low metabolic performance. It means, that no direct correlation exists between actual blood glucose value and cell metabolic state.
Relation Between Blood Glucose Values and Cell Metabolism in Repetitive Stress-Condition
Increasing metabolic performance under stress is possible as a result of the repeated application of the stress trigger (i.e., the stressor).
For example the transient metabolic deficit insults induced by the repeated physical training (so-called "metabolic training") lead to an increase in the activity and quantity of the enzyme cascades involved in glucose and free fatty acid uptake and in the glucose and free fatty aciddependent metabolic chain. All these alterations result in augmented functional performance [3,4,9,12,22]. It should be noted, that he augmented performance of cell metabolism is not reflected in blood glucose values.
In consideration of repetitive loading, substrate utilization changes in a typical manner: free fatty acid dominance at the expense of glucose comes about. To site an example, free fatty acid substrate dominance is a characteristic of allostatic load in organs which perform continuous work including the heart, the oxygen-enrich renal cortex and the liver [3,8,11,12,15,22].
The result of metabolic training is an increase in metabolic performance. Obviously, this adaptation is fundamentally different under various functional tasking’s.
For example, a sprinter who completes a distance in one single breath will meet his energy needs primarily from creatine phosphate stores and from adenosin triphophate synthesis produced by anaerobic glycolysis derived from glycogen stores. During this acute load, a significant oxygen debt is generated, which is characterized by a negative metabolic energy balance. This metabolic state is associated with allostatic overload. Therefore, the sprinter's metabolic training is predominantly aimed at increasing the functional performance in creatine phosphate and glycogen stores, as well as the enzyme chain of anaerobic glycolysis, while developing the necessary ultrastructure [3,7].
A marathon runner, on the other hand, is in a metabolic state of equilibrium throughout his or her workload. This is achieved by shifting substrate utilization predominantly to free fatty acid in preference to glucose, since this enables a far more beneficial adenisine triphosphate production (38 vs. 133) at the molecular level [3,7].
A further adaptive mechanism is an increase in myoglobin stores of striated muscle (the proliferation of "red muscle"), which provides a significant excess of oxygen supply [3].
The metabolic adaptation regarding a marathon runner involves both functional and morphological augmentation in the mitochondrial aerobic oxidation enzyme cascade, which is reflected in an increase in the number of mitochondria and a distinctive change in their morphology
It can also be seen while undergoing a prolonged elevated metabolic performance (i.e. hyper catabolic state) acute stress tolerance (the acutely inducible functional reserve performance) is reduced: the maximum performance of a marathon runner at the finish line must not be equal to that of a sprinter.
In repetitive stress, adaptation involves not only the increase in function associated with membrane receptors, but also ultra-structural and macro morphological alterations resulting from de novo protein synthesis associated with nuclear receptors [3,4,9].
It is also evident, following stressful workloads; the body's free energy level, the replenishment of depleted stores, and the reparation of ultra-structural damage are dependent on adequately long periods of rest and sufficient quantity and quality of calorie intake during 'inter-stress' periods. The harmony between the length of the inter-stress phases and the nutritional clusters is a prerequisite for full recovery. If this is reached, adaptation will have achieved its goal: the body will successfully adapt to the stressor, otherwise the body risks its health [3,7,8,21].
The endocrine messenger clusters of the stress phase and the inter-stress phase are fundamentally different, even opposite from one another: in contrast to the catabolic nature of the stress phase, the inter-stress phase is characterized by hyper insulinemia and the associated hypoglycemic tendency due to anabolic targeting [3,7,21]. In regards to stress phase catabolism, in addition to the dominance of stress hormones, minimal amounts of insulin are required to achieve maximal metabolic and functional performance [3,9].
Relation Between Blood Glucose Values and Cell Metabolism in Chronic Sress-Condition
A chronic stress state occurs when adaptation fails to achieve its goal, i.e., the stress trigger (the stressor) is persistent, causing the stress response to remain chronically active, threatening health of the organism.
The Pathophysiological, Metabolic Consequences are, as follows:
The stress trigger activates the stress hormones (catecholamine, glucagon, etc.,), which is necessarily accompanied by the increased metabolic and functional activity of the stress organs (heart, lungs, brain, etc.,) and a progressive dominance of free fatty acid use at the expense of glucose due to chronic hyper metabolism [1,3,13].
In addition to the involved stress organs, the entire body is affected by the following changes.
Chronic catabolic metabolic activation shows the opposite metabolic orientation to anabolic processes, i.e., the replenishment of nutrient stores and the occurrence of ultra-structural regeneration can only be achieved with certain limitations. The inevitable consequence of this is the inducible metabolic, the functional reserve capacity, and the acute stress tolerance is reduced.
The stress hormone-induced insulin resistance leads first to a tendency to hyperglycemia and next manifests as diabetes, which, in the lack of appropriate intervention, results in a self-exciting progressive process through the mechanism of glucose toxicity, in which both the insulin resistance and the hyperglycemia undergo an increase [3,4,10,11,13,16,23,24].
Despite compensatory hyper insulinemia, cellular metabolism can be characterized by insulin hypofunction. In this progressive dysmetabolism, the cellular metabolic balance is maintained for a limited period (allostatic load), however, over a span of time macroerg phosphate production is unable to compensate for its utilization: the metabolic balance is upset, a tendency to ketoacidosis and lactic acidosis appear due to the damage regarding mitochondrial processes (allostatic overload).
The progression of ultra-structural lesions is exacerbated by the increased oxygen free radical and calcium ion overload associated with hyper metabolism, which is accelerated when allostasis becomes disrupted. The aggravation of ultra-structural lesions leads to further deterioration in metabolic performance, resulting in a circulus vitiosus progression mechanism [1,3,12,13,15].
This process, known as metabolic remodeling, affects all cells in the organism, however, the metabolically active organs are most likely to be at risk of metabolic decompensating. Mainly, the continuously functioning organs (heart, vascular system) are exposed, which explains the leading position of cardiovascular risk growth.
Therefore, vital organs deserve special attention from the perspective of being both the causes and the sufferers in chronic stress.
In conclusion with respect to the above, acute and repetitive stress, so as long as it is able to avert or adapt to the presence of the stressor threatening the organism, can be considered a successful adaptation, which corresponds to Selye's concept of "good stress" (eustress). In contrast, chronic stress of decompensated metabolic phase should be considered a failed coping process, which is a progressive disease process which injures the body. This corresponds to Selye’s concept of the state of “bad stress” (distress). Again it should be emphasized, that two stages of chronic stress show diverse feature regarding metabolic balannce, which is not reflected in the actual blood glucose values (Figure 2).
The metabolic dysfunctional characteristic of the decompensated phase of chronic stress (distress) is illustrated by Hippocrates' statement, "nutrition is a medicine for the convalescent organism, a poison for the sick organism" [3]. The metabolic explanation for this observation implies the success of regenerative processes can only be ensured by nutrient intake once cell metabolism achieves balance, and, if cell metabolism is negative, regeneration cannot be achieved by nutrient intake.
The Relationship Between dysmetabolism, and Chronic Stress, Diabetes, Ischemic Damage
Chronic dysmetabolism in any vital organ, from any cause, generates a persistent stress trigger (stressor), which inevitably leads to chronic stress activation. It is well documented that psych mental discomfort (e.g., civilization stress) can induce the same metabolic, functional, ultra- -structural, micro-/macroscopic cellular and organ alterations as “substantial or material” stressors [1,3-6].
Euglycemic dysmetabolism (metabolic syndrome) inescapably results in persistent hyperglycemia over time [3,4,10,16]: i.e., causes type 2 diabetes, which can be explained by the chronic stress activation induced due to the primary metabolic dysregulation. This disordered cardiac metabolic condition of diabetic patients (i.e., the diabetic cardiomyopathy) acts as an independent risk factor, which per se is predisposed to the development of heart failure [25].
Chronic stress, as explained above, leads obligatorily to the development of diabetes and to the increased and prolonged presence of cardiovascular risk factors: the rise in serum cholesterol and triglyceride serum levels, the development of hypertension, well-documented hemostasis and inflammatory changes [1,13,23,26]. The consequent generalized endothelial dysfunction results in macro-, micro angiopathy, left ventricular hypertrophy, and then in clinical events of ischemic origin (coronary artery disease, stroke threat) [1,13,23,26].
Comprehensively, this means that chronic stress can be regarded as a pathogenetic factor in its own terms: diabetic, ischemic and chronic stress-induced dysmetabolism (pathogenesis see above) appear inseparably and together. All three components can be labeled as both cause and effect since it is a self-exciting process with chronic stress (distress) at its core.
The chronic stress - induced detrimental pathogenetic cascade results in organ failure of vital organs (Figure 3). Besides, insulin resistance developing other types of endocrine resistance is also associated with the evolution of chronic stress [27] (Figure 4).
Glucotoxicity is the dominant component of the pathogenetic process, which maintains the progression of metabolic decay [1,3,13,21,23,26,28].
On the other hand, it is well documented that in chronic pathological conditions of vital organs as chronic heart failure, coronary artery disease, chronic kidney disease, liver disease or lung disease diabetes inevitably develops, induced by the chronic stress-activation [1,3,13,29-31], moreover,the multifaceted clinical sequelae of chronic stress include obesity, metabolic syndrome, type 2 diabetes, hypertension, Alzheimer's disease, non-alcoholic fatty liver, and increased incidence of various malignancies [1,13,32,33].
The link between chronic stress and aging is tempting: the age-related decline in vital organs' metabolic performance could be a stress-triggering factor.
Relation Between Blood Glucose Values and Cell Metabolism in Diabetes
Acute blood glucose elevation is a consequence of acute stress, which is part of the self-healing metabolic adaptation. During the allostatic load phase, the hyperglycemia of acute stress results in increased metabolic performance, thus indicates eumetabolism, i.e., it is deemed beneficial [2,3,7,14].On the contrary, in allostatic overload, hyperglycemia is already a feature of dysmetabolism, i.e., it can be considered a pathological parameter. This suggests the compensated phase (eustress) and in the decompensated phase (distress) of acute stress hyperglycemia is linked to an opposite metabolic performance.
In a chronic hyperglycemic state (diabetes), acute stress-associated sharp blood glucose elevation can theoretically be regarded as a transient, advantageous metabolic phenomenon.
Persistent hyperglycemia (diabetes) is always considered to be an adverse pathological factor. On the one hand, chronic blood glucose elevation is a "poison" to all cells (glucose poisoning) [1,3,14,23,26,34,35], and becomes a vicious circle which constantly deteriorates itself through the mechanism of glucotoxicity. This phenomenon can be defined as the "maker" function of chronic hyperglycemia ("glucotoxicity-induced dysmetabolism").
Consequently, glucotoxicity is essentially a specific phenomenon of glucose-poisoning, i.e., it can be understood as a "glucose-poisoning" specific in terms of the target: i.e., beta-cells, hepatic glucose outflow and peripheral insulin resistance.
However, insulin treatment can only be considered as a causal intervention in insulin-deficient type 1 diabetes and it can be regarded as a symptomatic treatment in other forms of diabetes.
In chronic stress, the elimination of the stressor should be viewed as the causal therapy [36,37]. Possibilities associated with the treatment of chronic stress are: “stress relief techniques” (psych mental approach), dietary, life-style interventions, pharmacological treatment (reversal of metabolic remodeling) [36,38].
Moreover, chronic hyperglycemia is referred to not only a "maker" but also a "marker". Namely, it is a mandatory indicator of the prolonged presence of a cellular metabolic disorder as a primary stress trigger and the existence of a consequent chronic stress activation, for example, the transition from metabolic syndrome to type 2 diabetes [3,4,10,16].This implies chronic hyperglycemia can be referred to as both a ‘maker’, and a cause, and also, as a 'marker', a consequence.
Therefore, it is evident that near-normoglycemia, as a primary focus in the treatment of diabetes, is clearly a well-grounded thesis, since its implementation disables, or at least moderates, the glucotoxicity mechanism, which is the primary factor in progression of metabolic deterioration, i.e., the primary trigger in the maintenance of chronic stress. It is important to emphasize that blood glucose value is of crucial importance in governing the therapy of diabetes, but not in the assessment of the cellular metabolic state.
A large number of “metabolic promoter” drugs are proposed to exert their beneficial emetabolic effect on cell metabolism by “blood glucose independent way” [39-41].
It is well known from the results of euglycaemic clamp studies that the simultaneous administration of various amounts of intravenous glucose and appropriate insulin dosages result in permanently normal blood glucose values. It is obvious that the identical blood glucose values observed during the euglycaemic clamp do not reveal either the significantly various degrees of glucose disposal or the extremely different metabolic activities of tissues.
The statement that there is no strict association between blood glucose values and cell metabolism, furthermore, euglycaemia does not necessarily indicate eumetabolism supported by several clinical observations and trials: UK Prospective Diabetes Study Group, CREATE ECLA Trial,Saint Antonio Heart Study, the GUSTO-I experience [25,42-46].
Metabolic memory explained by the preexisting intensified insulin treatment leading to persistently improved cell metabolism (“legacy effect”), could not be demonstrated by the post existing blood glucose values compared to those of controls with similar blood glucose range, but without metabolic memory [47]. It means, that blood glucose values do not reflect the actual cellular metabolic state.
In summary, it can be concluded that blood glucose levels used as an approach for characterizing cellular metabolism is no longer established. The rationale behind this statement is that blood glucose can really be referred to a transport parameter, which reflects the actual equilibrium between glucose transport to the blood from the intestinal tract and glucose deposits, on the one hand, and glucose transport into peripheral tissues (glucose disposal) from the blood, on the other hand. For this reason, blood glucose values cannot provide real qualitative and quantitative information about the characteristics of cell metabolism neither in a defined nor in different organs [1].
Relation Between Morphologic and Metabolic Adaptation in Stress Condition
Changes in the external and internal environment of the organism, characterized as triggers, induce adaptation responses (e.g., stress response) in the organism. Two components of this adaptation can be distinguished: there is a somato-morphological adaptation and there is a metabolic adaptation. A significant difference between the two is that morphological adaptation is always trigger-specific, whereas metabolic adaptation is not stressor-specific (Figure 5). Of the two mechanisms, from a physiological aspect, metabolic adaptation is considered to be the dominant one, since the increase in metabolic performance provides the prerequisites for functional, ultra-structural and macro morphological changes in organs.
Specific somato-morphological adaptation is characterized by the often referred to phenotypic plasticity, which is based on genetic and epigenetic mechanisms [3,4,9,48]. In analogy of a metaphor: the string spectrum of the piano symbolizes the inherited gene spectrum, the pianist symbolizes epigenetics, and the sheet music symbolizes environmental stimuli. Next, the pianist (epigenetics) plays a different melody (phenotype) depending on the variation of the music score (i.e., external or internal environmental trigger), in which the melody (phenotype) played will reflectthe score to a varying degree depending on the talent of the pianist (epigenetics) and the piano's endowment (genome) [1,3,16,48].
As a result, the organism reflects changes in its external or internal environment by phenotype modification. An extreme result of this can be, from one perspective, when identical twins develop significantly different phenotypes due to fundamental differences in the external environment, despite the fact they share the same genome. From yet another perspective, genetically unrelated individuals may respond to one another by developing similar phenotypes due to similarities in the external or internal environment [49-53]. Trigger-specific afferent information is translated into an efferent genetic language by the autonomic neuro-endocrine network connecting all cells: trigger-specific codes are activated from the genome, resulting in trigger-specific de novo protein synthesis, trigger-specific ultra- -structural and macro morphological changes, and the development of a trigger-specific phenotype [49,50].
These results in a large number of phenotypes of a large number of pathological entities (pathological states) induced by extremely heterogeneous pathogenetic pathogens (triggers), and in a clear similarity between phenotypes of the same cause, i.e., the phenotype is always trigger-specific: thyroid patients, renal patients, liver patients and tobacco users all bear a typical phenotype [49-53].
In contrast, in Selye's stress response, metabolic adaptation is not stressor-specific: i.e., it can only respond in the same way to any trigger. The spectrum of potential manifestations is limited: acute stress, repetitive stress and chronic stress including allostatic and allostatic overload forms.
An environmental trigger becomes a stressor (i.e., a stress activator) once the triggered process causes damage to the functionality regarding a vital organ, thus activating a non-specific rescue response of the body, defined as Selye's stress response. The metabolic characteristics of stress response are determined by both genetic (thrifty genotype) and epigenetic (thrifty phenotype) factors [47].
The above stress states can be well characterized by the features of primary messengers, blood glucose and free fatty acid levels, cellular substrate uptake, anaerobic glycolysis, mitochondrial aerobic oxidation, ketosis disorder, inducible metabolic and functional capacity and acute stress tolerance.
On the basis of these characteristics, it is theoretically possible to identify these distinct stress-associated metabolic states [4,9,11,19,54-56]. The clinical methodology of this approach is not available in practice, although the proposed "metabolic diagnosis" could provide a more accurate insight into the body's cellular metabolism than the blood glucose level.
rate insight into the body's cellular metabolism than the blood glucose level.
If we consider the patterns of metabolic adaptation, we can draw parallels between pregnancy, starvation and SGLT2 inhibitor treatment.
All of these conditions are characterized by high free fatty acid serum levels, the dominance of free fatty acid substrate utilization at the expense of glucose, reduced inducible functional performance and stress tolerance, acetonemic tendency and impaired carbohydrate metabolism.
All these parameters correspond to a state of chronic stress in which catabolic metabolism, reduced acute stress tolerance, is already present while the energetic balance of cellular metabolism is preserved.
A higher frequency of post-operative euglycemic diabetic ketoacidosis, hyperglucagonemia, hypoinsulinemia and, surprisingly, reduced physical performance have been documented in patients treated with SGLT2 inhibitors [50,57,58].
Gliflozin-induced glucosuria can be paralleled with the glucose-lowering effect of the commonly referred to fetal siphon-effect during pregnancy. Both conditions are associated with relative starvation. The common stressor in starvation, during pregnancy, and in SGLT2 inhibitor treatment is glucose deprivation, the presence of which activates non-specific stress adaptation [4,7,31,36,57-59].
Gliflozin-induced glucosuria can be paralleled with the glucose-lowering effect of the commonly referred to fetal siphon-effect during pregnancy. Both conditions are associated with relative starvation. The common stressor in starvation, during pregnancy, and in SGLT2 inhibitor treatment is glucose deprivation, the presence of which activates non-specific stress adaptation [4,7,31,36,57-59].
The cardio protective effect of SGLT2 inhibitors may be explained by several factors.
The moderation of glucotoxicity, which is considered a crucial deteriorating component in complex self-exciting pathophysiological processes of chronic stress.
In mild ketosis induced by SGLT2 inhibitors, the production of ketone bodies is enhanced. Increased aerobic oxidation of beta-oxybutyric acid has been associated with the so-called off-label metabolic effect induced by gliflozine treatment. This mechanism is independent of the SGLT-2 protein and is characterized by an exceptionally beneficial metabolic efficiency [60-62].
On the basis of similarities between pregnancy, and starvation, and SGLT2 treatment these states can be referred to as a “glucopenic stress-ketosis”, which can be characterized by an optimal metabolic efficiency. It is important to note, that although in glucopenic stress-ketosis the metabolic performance is outstandingly beneficial, but the acute stress tolerance is diminished in association with decreased inducible reserve metabolic capacity supported by permanent katabolic activity [60,63,64].
As all the pathological states are induced by cellular metabolic disorder of different origin and organ location, therefore a general metabolic promoter can beneficially act as a non-specific symptomatic intervention. This supposition can theoretically extend the indication spectrum of SGLT-2 inhibitors.
In addition to the general metabolic promotion described above, the Reno protection is also explained by the suspension of high-energy glucose transport in the proximal tubules, which results in the maintenance of cellular energy balance, hence, the improvement of other renal functions. This effect is apparently independent of blood glucose values [60-62].
Conclusions
Metabolism is the most basic feature of the living organism. Therefore, the availability of a method to assess the metabolic state theoretically is a fundamental requirement.
Blood glucose value is not appropriate in characterizing the cell metabolism: "blood glucose as a metabolic parameter" is not a sustainable thesis.
"Near-normoglycemia" as a therapeutic target based on blood glucose measurement is a mandatory thesis in management of the diabetes therapy
The somato-morphological adaptation induced by changes in the external/internal environment of the organism (the presence of a trigger) is always trigger-specific, i.e., a large number of heterogeneous triggers indicates the large number of phenotypes which are evolved and differ from one another.
In stressor-activated stress responses (acute, repetitive, and chronic stress states), metabolic adaptation is not stressor-specific, i.e., regardless of a large number of heterogeneous causes, the metabolic display is limited to a narrow spectrum.
In accordance with the law of "all or nothing”, in reference to acute stress, the body either eliminates the stressor or the body inevitably becomes its victim.
The repeated, transient presence of a stressor (repetitive stress) can lead to an increase in the functional performance of the organism by creating a simultaneous morphological adaptation ("constructive" eustress). The success of adaptation to a stressor depends on the duration and nutritional properties of the so-called inter-stress periods.
Chronic stress disorder with negative energetic balance is a self-generating; progressive, harmful process ("destructive" distress) induced by constant presence of the stressor.
The “gulucopenic stress-ketosis” of chronic stress associated with preserved energetic balance of cell metabolism can be characterized by an optimal metabolic efficiency
The distinct metabolic characteristics of acute, repetitive, and chronic stress reactions can be well described and their identification (the "metabolic diagnosis") provides a more accurate approach in defining the metabolic state of the organism compared to determination of blood glucose values.
- Simon K, Wittmann I (2019) Can blood glucose value really be referred to as a metabolik parameter? Rev Endocr Metab Disord, 20: 151-60.
- Selye H (1936) A Syndrome produced by diverse nocuous agents. Nature, 138: 32.
- El Bacha T, Luz M, Da Poian A (2010) Dynamic adaptation of nutrient utilization in humans. Nat Educ, 3: 8.
- Picard M, McEwen BS, Epel ES (2018) An energetic view of stress: focus on mitochondria. Front Neuro endocrinol, 49: 72-85.
- Tattersall RB (1989) Type 2 diabetes or NIDDM: looking for a better name. Lancet 1: 589-91.
- Taskinen MR (2000) Lipid metabolism in diabetes. In: Taskinen R, editor. Diabetes int the new millenium. Philadelphia: W.B. Saunders Company, 32-55.
- Brun JF, Dumortier M, Fedou C, et al. (2001) Exercise hypoglycemia in nondiabetic subjects. Diabetes Metab, 27: 92-106.
- Turner N (2013) Mitochondrial metabolism and insulin action.
- Karakelides H, Asmann YW, Bigelow ML, et al. (2007) Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes, 56: 2683-9.
- Boden G (2003) Effects of free fatty acids (FFA) on glucose metabolism: significance for insulin resistance and type 2 diabetes. Exp Clin Endocrinol Diabetes, 111: 121-4.
- Groop LC, Bonadonna RC, DelPrato S, et al. (1989) Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest, 84: 205-13.
- Ward SA. Rossiter HB (2004) Physiology of exercise. In: Principles of Exercise Testing and Interpretation, K Wasserman, J Hansen, D Sue, W Stringer, B Whipp, eds. Lippincott Williams & Wilkins, Philadelphia, 8-58.
- Wilcox G (2005) Insulin and insulin resistance. Clin Biochem Rev, 26: 19-39.
- Selye H (1974) Stress without distress. Philadephia, PA: JB. Lippincott.
- Kassiotis C, Rajabi M, Taegtmeyer H (2008) Metabolic reserve of the heart: the forgotten link between contraction and coronary flow. Prog Cardiovasc Dis, 51: 74-88.
- Abel ED (2010) Free fatty acid oxidation in insulin resistance and obesity. Heart Metab, 48: 5-10.
- Barmore W, Patel H, Harrell S, Garcia D, Calkins Jr JB. (2022) Takotsubo cardiomyopathy: A comprehensive review. World J Cardiol, 14: 355-62.
- Bell DS (2004) Advantages of a third-generation beta-blocker in patients with diabetes mellitus. Am J Cardiol, 93: 49B-52.
- Wan T, Ge ZD, Tampo A, et al. (2008) The A3 adenosine receptor agonist CP-532,903 (N6-(2,5 dichlorobenzyl)-3-aminoadenosine-5-N- methylcarboxamide) protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J Pharmacol Exp Ther, 324: 234-43.
- Zhou G, Myers R, Li Y, et al. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest, 108: 1167-74.
- Shaw E, Leung G, Jong J, et al. (2019) The impact of time of day on energy expenditure: implications for longterm energy balance. Nutrients, 11: 2383.
- Berg J, Tymoczko J, Stryer L (2002) Biochemistry. New York, NY: WH Freeman.
- Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest, 106: 473-81.
- Ceriello A (1998) The emerging role of post-prandial hyperglycaemic spikes in the pathogenesis of diabetic complications. Diabet Med, 15: 188-93.
- Woodfield SL, Lundergan CF, Reiner JS, Greenhouse SW, et al. (1996)Angiographic findings and outcome in diabetic patients treated with thrombolytic therapy for acute myocardial infarction: the GUSTO-I experience. J Am Coll Cardiol, 28: 1661-9.
- Nathan DM, Genuth S, Lachin J, et al. (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med, 329: 977-86.
- Wittmann I (2023) The Common Single Cause of Chronic Multi-Hormonal Resistance in Oxidative Stress. Antioxidants, 12: 75.
- DeFronzo RA (1992) Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia, 35: 389-97.
- Taegtmeyer H (2004) Cardiac metabolism as a target for the treatment of heart failure. Circulation, 110: 894-96.
- Guha A, Harmancey R, Taegtmeyer H (2008) Nonischemic heart failure in diabetes mellitus. Curr Opin Cardiol, 23: 241-8.
- Xiaofang Y, Zhang S, Zang L (2018) Newer perspecties of mechanisms for euglycemic diabetic ketoacidosis. International Journal of Endocrinol, 2018: 7074868.
- Stern MP (1995) Diabetes and cardiovascular disease: the "common soil" hypothesis. Diabetes 44: 369-74.
- Dinneen S, Gerich J, Rizza R (1992) Carbohydrate metabolism in non-insulin-dependent diabetes mellitus. N Engl J Med, 327: 707-13.
- van der Berghe G (2005) Insulin vs. strict blood glucose control to achieve a survival benefit after AMI? Eur Heart J, 26: 639-41.
- Koutroumpakis E, Jozwik B, Aguilar D, et al. (2020) Strategies of Unloading the Failing Heart from Metabolic Stress. Am J Med, 133: 290-6.
- Miller MJ, Kutcher J, Adams KL (2017) Effect of pregnancy on performance of a standardized physical fitness test. Mil Med, 182: 1859-63.
- Haffner SM (2006) Abdominal obesity, insulin resistance, and cardiovascular risk in pre-diabetes and type 2 diabetes. Eur Heart J, 20-5.
- Sinclair KD, Allegrucci C, Singh R et al. (2007) DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc. Natl. Acad. Sci. USA, 104: 19351-6.
- Krentz AJ, Bailey CJ (2005) Oral antidiabetic agents: current role in type 2 diabetes mellitus. Drugs, 65: 385-411.
- Hanefeld M, Cagatay M, Petrowitsch T, Neuser D, Petzinna D, Rupp M (2004) Acarbose reduces the risk for myocardial infarction in type 2 diabetic patients: meta-analysis of seven long-term studies. Eur Heart J, 25: 10-6.
- Adler AI (2001) Cardiovascular risk reduction in diabetes: underemphasised and overdue. Messages from major trials. Clin Med, 1: 472-7.
- UK Prospective Diabetes Study (UKPDS) Group (1998) Intensive bloodglucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group Lancet, 352: 837-53.
- UK Prospective Diabetes Study (UKPDS) Group (1998) Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group Lancet, 352: 854-65.
- Mehta SR, Yusuf S, Diaz R, et al. (2005) Create Ecla trial group investigators. Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction: the CREATE-ECLA randomized controlled trial. JAMA, 293: 437-46.
- Apstein CS, Opie LH (2005) A challenge to the metabolic approach to myocardial ischaemia. Eur Heart J, 26: 956-9.
- Hunt KJ, Resendez RG, Williams K, Haffner SM, Stern MP, Heart SA (2004) National Cholesterol Education Program versus World Health Organization metabolic syndrome in relation to all-cause and cardiovascular mortality in the San Antonio Heart study. Circulation, 110: 1251-7.
- LeRoith D, Fronseca V, Vinik A (2005) Metabolic memory in diabetes – focus on insulin. Diabetes Metab Res Rev, 21: 85-90.
- Simon K (2012) Chronic stress and epigenetics. [Krónikus stressz és epigenetika.] Orv Hetil, 153: 525-30.
- Simon K (2015) Diagnosis based on the patient phenotype. Hung Med J, 156: 1144-51.
- Simon K, Gyulai M, Tamas G, et al. (1999) Screening for type II diabetes-candidates. Am J Med, 106: 489-91.
- Feinberg AP (2007) Phenotypic plasticity and the epigenetics of human disease, 447: 433-40.
- Johnstone SE, Baylin SB (2010) Stress and the epigenetic landscape: a link to the pathobiology of human diseases? Nat Rev Neurosci, 11: 806-12.
- Humphrey G (1924) The psychology of the Gestalt. J Educ Psychology, 15: 401-12.
- Sharma S, Singh H, Ahmad N, et al. (2015) The role of melatonin in diabetes: therapeutic implications. Arch Endocrinol Metab, 59: 391-9.
- Simon K (2021) Stress characterization is a more reliable approach for the assessment of metabolism than blood glucose value. J Diabet Res Rev Rep, 3: 1-7.
- Simon K, Wittmann I (2022) More reliable approach than blood glucose value is needed in assessment of cell metabolism. Chapter 3 eBook ISBN: 978-93-5547-712-5, 10: 973/6pi/codhr/v2/2265A.
- Linden MA, Ross TT, Beebe DA, et al. (2019) The combination of exercise training and sodium-glucose cotransporter-2 inhibition improves glucose tolerance and exercise capacity in a rodent model of type 2 diabetes. Metabolism, 97: 68-80.
- Newman AA, Grimm NC, Wilburn JR, et al. (2019) Influence of Sodium Glucose Cotransporter 2 Inhibition on Physiological Adaptation to Endurance Exercise Training, 104: 1953-66.
- Woodfield SL, Lundergan CF, Reiner JS, et al. (1886) Angiographic findings and outcome in diabetic patients treated with thrombolytic therapy for acute myocardial infarction: the GUSTO-I experience. J Am Coll Cardiol, 28: 1661-9.
- Buse JB, Wexler DJ, Tsapas A, et al. (2020) 2019 update to: Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia, 63: 221-8
- Tentolouris A, Vlachakis P, Tzeravini, et al. (2019) SGLT2 inhibitors: a review of their antidiabetic and cardioprotective effects. Int J Environ Res Public Health, 16: 2965.
- Dyck JR, Sossala S, Hamdani N, et al. (2022) Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: evidence for potential off-target effects. J Mol Cell Cardiol, 167: 17-31.
- Selvaraj S, Kelly, DP, Margulies KB (2020) Implications of Altered Ketone Metabolism and Therapeutic Ketosis in Heart Failure. Circulation, 141: 1800-12.
- Kolb H, Kempf K,Röhling M, Lenzen-Schulte M, Nanette C, Schloot 1, Martin S (2021) Ketone bodies: from enemy to friend and guardian angel. BMC Medicine, 19: 313.
FIGURE 1
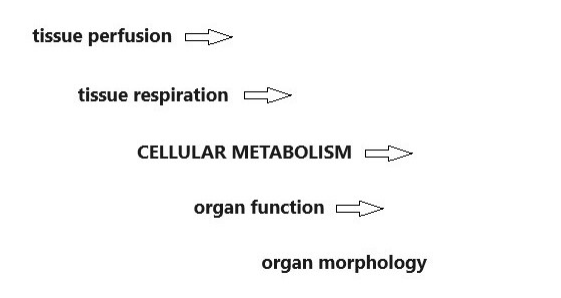
Figure 1: Central position of metabolism in the organism
FIGURE 2
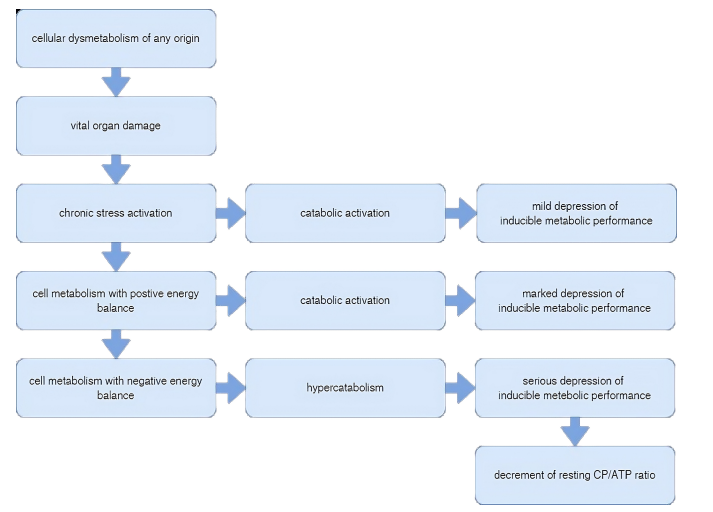
Figure 2: Metabolic dysregulation in chronic stress
FIGURE 3
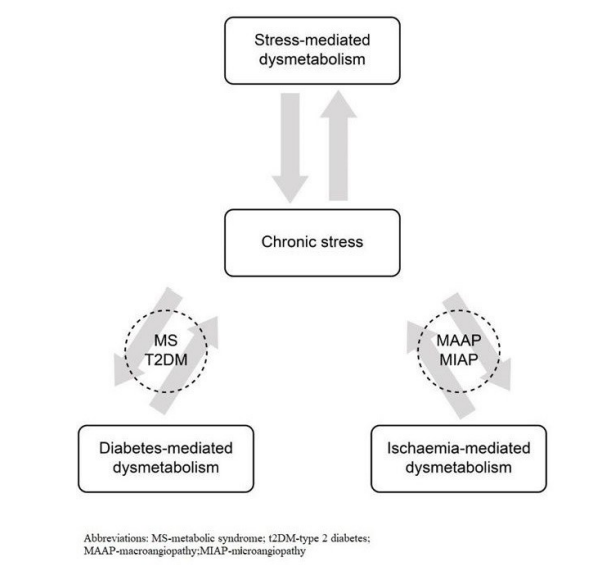
Figure 3: Interrelation between chronic stress-, and diabetes-, and ischaemia-induced dysmetabolism
FIGURE 4
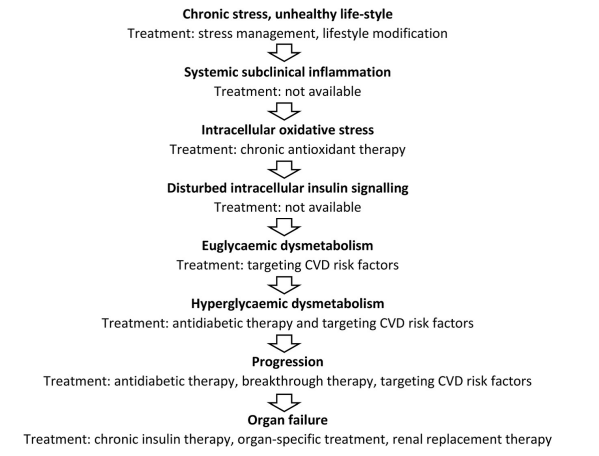
Figure 4: Pathogenesis of chronic stress-induced diabetic dysmetabolism
FIGURE 5
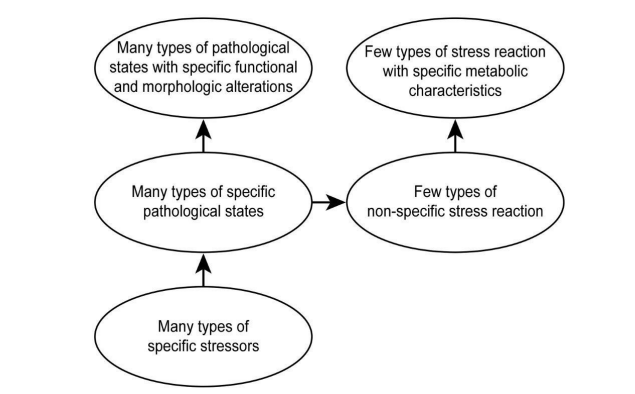
Figure 5: Specific morphologic and non-specific metabolic adaptation in stress reaction
Figures at a glance