Density Functional Theory Study of Structural, Electronic, Elastic, Phonon, Thermoelectric, and Ab Initio Molecular Dynamics Properties of MgX (X = O, S, Se, and Te)
Received Date: May 26, 2026 Accepted Date: June 07, 2026 Published Date: June 12, 2026
doi:10.17303/jmsa.2026.10.103
Citation: Mazhar Haleem Awan, Sehrish Munsif (2026) Density Functional Theory Study of Structural, Electronic, Elastic, Phonon, Thermoelectric, and Ab Initio Molecular Dynamics Properties of MgX (X = O, S, Se, and Te). J Mater sci Appl 10: 1-12
Abstract
Chalcogenide compounds have gained significant attention as versatile materials due to their exceptional capabilities in optoelectronics, catalysis, and renewable energy. This study comprehensively explores the structural, electronic, elastic, phonon, thermoelectric, and ab initio molecular dynamics (AIMD) properties of MgX compounds (where X = O, S, Se, and Te) using density functional theory (DFT) via the full-potential linearized augmented plane wave plus local orbital (FP-LAPW+lo) method. Their stability in the rock salt structure with space group (Fm¯3m symmetry) is confirmed by the computed lattice parameters for MgO (4.2616 Å), MgS (5.2411 Å), MgSe (5.5153 Å) and MgTe (5.9783 Å). Electronic band structure calculations show that MgTe behaves as a semiconductor, featuring an indirect band gap of 0.171 eV when spin-orbit coupling (SOC) effects are taken into account. Elastic constants (C_11, C_12, and C_44) satisfy the mechanical stability criteria evaluated using the Born criterion. Phonon dispersion curves and AIMD simulations confirm the dynamical and thermal stability of the MgTe compound. The results indicate that the MgTe binary compound holds considerable promise for thermoelectric device applications, demonstrating high thermoelectric performance with ZT values of 0.96 for p-type carriers and 0.95 for n-type carriers, accompanied by power factor (PF) values of 48.72 and 6.61 (in arbitrary units consistent with the computational framework). These ZT values were estimated assuming a con- stant relaxation time approximation and a carrier concentration of approximately 10^19 to 10^20 cm^3, with electronic thermal conductivity dominating over lattice con- tributions under the considered doping levels.
Keywords: Density Functional Theory; Elastic Constants; AIMD; Phonon; Thermoelectric
Introduction
Binary alkaline-earth chalcogenides have recently gained significant interest due to their valuable physical properties and potential applications in optoelectronic and spintronic devices. Within this group, magnesium chalcogenide compounds, represented by the for- mula MgX (where X = O, S, Se, and Te), are part of the binary II-VI compound family. Among them, MgO stands out as the most ionic and insulating material, while MgS, MgSe, and MgTe act as semiconductors with decreasing ionicity in that order. These materials exhibit adjustable band gaps, stable crystal structures, and notable elastic and vibrational characteristics that are highly influenced by their elemental composition [1- 5]. Chalcogenide and related materials have been the subject of extensive theoretical and experimental research in recent years due to their remarkable structural versatility and composition-dependent physical behavior. Alkali and alkaline-earth metal chalco- genides, in particular, have emerged as chemically stable and environmentally benign substitutes for traditional semiconductor materials. Wide band gaps, good mechanical stability, and favorable optical responses have been shown in density functional theory (DFT) studies on compounds such as NaMgX3 and LiMgX3, indicating their suitabil- ity for ultraviolet (UV) optoelectronic applications [6, 7]. Theoretical studies on CaMgX3 compounds have similarly revealed mechanically stable structures with promising thermal and electronic properties. These results suggest that Mg-based chalcogenides represent an important class of materials for investigating novel multifunctional properties. How- ever, it is important to note that the present study focuses on binary MgX compounds with a rock salt structure, not on perovskite-type ternary oxides. Recent advances in first-principles calculations have unveiled intriguing electronic and magnetic properties in various oxide and chalcogenide materials. Compounds such as PrMnO3, BaFeO3, and CaFeO3 have been theoretically predicted to exhibit half-metallic behavior under equilibrium or strain-induced conditions. Notably, strain engineering and electron corre- lation effects have been shown to substantially alter the magnetic and electronic states of BaFeO3 and CaFeO3, resulting in half-metallic ferromagnetism and phase transitions [8-10]. Furthermore, studies using the modified Becke-Johnson potential combined with the Hubbard U correction have reported half-metallic ferromagnetism in BaNpO3 and related compounds. Similar behavior has also been observed in transition-metal-free ox- ides like LiBeO3 and KMgO3, where spin polarization primarily arises from p-orbital interactions rather than conventional d-electron magnetism [11-13]. Unlike conventional half-metallic ferromagnets based on transition metals, lightweight alkaline-earth com- pounds have recently emerged as promising candidates for spintronic applications due to their low toxicity, lower cost, and natural abundance. Specifically, KMgO3 has been theoretically predicted to exhibit thermodynamic stability in the ferromagnetic phase, along with a relatively high Curie temperature and strong magnetic properties stemming from oxygen p-orbital polarization [14]. These advancements indicate that alkaline-earth chalcogenides and related compounds offer a versatile platform for designing multifunc- tional materials with coupled electronic, magnetic, and thermal properties. Despite these significant advances, comprehensive studies that integrate structural stability, electronic structure, elastic response, phonon dispersion, thermoelectric performance, and thermal stability via AIMD simulations remain limited for binary MgX (X = O, S, Se, and Te) compounds. In particular, the systematic impact of chalcogen substitution on the elec- tronic, mechanical, vibrational, and thermodynamic properties of MgX materials is not yet fully understood. Moreover, previous research has seldom addressed the intercon- nected relationship between phonon stability, elastic properties, and finite-temperature thermal resilience-factors that are crucial for assessing the practical applicability of these materials in real-world device environments [15-17]. Therefore, this work aims to deliver a comprehensive first-principles investigation of MgX (X = O, S, Se, and Te) binary compounds using DFT. Structural, electronic, elastic, phonon, thermoelectric, and ther- mal stability properties are systematically analyzed to assess their potential for future optoelectronic and thermoelectric applications.
Computational Methods
All density functional theory (DFT) calculations were performed using the WIEN2k code [18] based on the full-potential linearized augmented plane wave plus local or- bitals (FP-LAPW+lo) method [19]. The exchange-correlation potential was treated using the Perdew-Burke-Ernzerhof generalized gradient approximation (PBE-GGA) [20]. For MgTe, spin-orbit coupling (SOC) was included within the PBE+SOC-GGA framework [20] to accurately capture the electronic structure and thermoelectric properties. The plane wave cutoff parameter was defined by RMT×KMAX = 7.0, where RMT is the smallest muffin-tin radius and KMAX is the maximum magnitude of the reciprocal lat- tice vector. The muffin-tin radii were set to 2.37 a.u. for Mg and 2.50 a.u. for O, S, Se, and Te. The maximum angular momentum for partial waves inside the spheres was lmax = 12, and the charge density Fourier expansion cutoff was Gmax = 14 au−1. A 14 × 14 × 14 k-mesh was used for self-consistent field calculations, and a denser 16 × 16 × 16 mesh was used for accurate electronic property evaluation. The energy convergence criterion was set to 10−5 Ry, and tetrahedral smearing with a broadening parameter of 0.0010 Ry was applied. Elastic constants (C11, C12, and C44) were calculated using the energy-strain method implemented in the WIEN2k code [21, 22], wherein cubic lattice deformations were applied and the resulting energy changes were fitted to second-order strain coefficients. Mechanical stability was verified using the born stability criteria [26]. Phonon dispersion curves were computed using the PHONOPY code [29] within the Parlinski-Li-Kawazoe supercell approach. A 2 × 1 × 1 supercell was constructed, and the force constants were obtained from DFT calculations using a 1 × 1 × 4 q-mesh. Thermoelectric properties, including the Seebeck coefficient (S), power factor (PF), and figure of merit (ZT), were calculated using the BoltzTraP code [27] based on the constant relaxation time approx- imation (CRTA). A dense k-mesh of 14 × 14 × 14 was used to ensure convergence of the transport integrals. Within the CRTA framework, ZT is expressed as ZT = S2σ T/ κe + κl, where σ is the electrical conductivity, κe is the electronic thermal conductivity, and κl is the lattice thermal conductivity. In this study, κl was not explicitly calculated; therefore, the reported ZT values represent upper bounds based solely on electronic con- tributions (κl = 0). A carrier concentration of 5 × 1019 cm−3 was assumed for both p-type and n-type doping, consistent with typical doping levels for thermoelectric materials [28]. The relaxation time (τ ) was treated as constant (CRTA) and cancels out in the ZT expression, as it appears in both the numerator and denominator under the ap- proximation that τ is energy-independent. Consequently, the absolute values of electrical conductivity and electronic thermal conductivity are not required independently; only their ratio matters for ZT within this approximation. Structural optimizations and total energy calculations were fitted to the Birch-Murnaghan equation of state [25]. Ab initio molecular dynamics (AIMD) simulations were performed using the WIEN2k molecular dynamics module to assess thermal stability. The simulations were carried out at 300 K for 12 ps with a time step of 1 fs. The Nose-Hoover thermostat [23] was employed to maintain constant temperature. Structural visualizations were generated using VESTA software [24].
Results and Discussions
Structural Properties
The compounds with the formula MgX (where X = O, S, Se and Te) are binary alkaline- earth metal chalcogenide compounds. They crystallize in the rock salt structure with the space group of Fm¯3m (No. 225). In the unit cell, the Mg atom occupies the 1-a Wyckoff position at fractional coordinates (0, 0, 0), located at the cube corners. The chalcogen atom X is at the 1-b site with coordinates (0.5, 0.5, 0.5), situated at the body center of the cubic lattice. This arrangement forms the ideal rock salt binary compounds-type MgX structural framework. By VESTA software, Figure 1 (a, b, c and d) shows that two atoms are placed in different arrangements of the elements. The structural properties of MgX (X = O, S, Se and Te) were studied by optimizing their lattice constants. The energy-volume (E-V) curves for MgO, MgS, MgSe, and MgTe were generated using the Birch-Murnaghan equation of state (EOS) [32], which confirmed the structural stability of these compounds [30-33]. In Figure 2(a-b), first optimization process is done by using DFT software to obtaining their different minimal energies: MgO (-551.43 Ryd) black, MgS (-1199.27 Ryd) red, MgSe (-5261.32 Ryd) green and last one MgTe (-13994.59 Ryd) blue. After these analysis MgTe compound become most stable element as compared to other one. Here the most appropriate potential is use to be PBE-GGA approximation to finding out minimized forces, and the system was unfastened until the forces acting on each atoms of the unit cell were negligible. Table 1 is obtained by Equation of Birch- Murnaghan (EOS) [32], which reveals that all calculated values of lattice constants that are in good agreement with other available data [33]. Equation 1 includes B0, B , V0, and E(V), which are the bulk modulus, the first derivative of unit cell volume, and energy at equilibrium ground states, respectively.
Band Structure and Thermoelectric Properties
Electronic band structure analysis offers essential insights into the physical properties of solids. It allows for the classification of materials as metallic or semiconducting by examining their characteristic electronic features. Band structure can be find-out by PBE-GGA exchange correlation potential method along with different potential in Table 2. According to the high symmetry orientations, i.e W, L, Γ, X, W, K points in the first brillioun zone (BZ) for rock salt structure of binary compound MgTe. Figure 3 displays the band structure for MgTe compound with different approximation are used to determining the bandgap values. The calculated band gaps reveal that the highest values are obtained using the HSE06 method, while the lowest values are predicted when spin-orbit coupling (SOC) is included. This study focuses on the narrow band gap, which enables rapid electronic transitions from the valence to the conduction band. Notably, MgTe exhibits an indirect band gap between the high-symmetry points Γ and X, with a calculated value of 0.171 eV. The obtained band gap, which narrows due to spin-orbit coupling, is beneficial for thermoelectric applications. The calculated figure of merit (ZT) and power factor (PF) demonstrate improved thermoelectric performance. Additionally, the band gap falls within an appropriate range for potential solar cell applications [34-35]. As shown in Table 3, the ZT values are 0.96 for p-type and 0.95 for n-type conduction, aligning well with previously reported literature values.
Phonon Dispersion Curves and AIMD Simulations
To verify the dynamical stability of MgTe in the rock salt structure with space group Fm¯3m phase, the phonon dispersion spectrum was analyzed, as presented in Figure 5. The calculated phonon band structure shows no imaginary (negative) phonon frequen- cies along the high-symmetry directions of the Brillouin zone, confirming the dynamical stability of MgTe in its rock salt structure. Since the primitive unit cell of MgTe contains two atoms, the phonon spectrum consists of three phonon branches, including two acous- tic branches and one optical branch. The acoustic branches originate from the Γ-point with zero frequency, satisfying the translational invariance condition, whereas the optical branch appears at higher frequencies due to the vibrational interactions between Mg and Te atoms. The absence of soft phonon modes further confirms that the crystal structure is dynamically and mechanically stable against lattice distortions.
Elastic Properties
When describing the materials in practical applications, efficient elastic constants are a helpful tool. The material’s reactivity to external forces and structural stability are characterized by its elastic properties. Three distinct components make up the elastic stiffness tensor for binary MgX (X = O, S, Se and Te) compounds with cubic symmetry: C11, C12, and C44 [36-37]. The calculated values of elastic constants for chalcogenide compounds are given in Table 4. The Born stability criteria, sometimes referred to as the mechanical stability conditions for these materials, must be met by the elastic stable compound MgX (X = O, S, Se and Te) [38-40]. It’s clear from Table 4 that, the computationally calculated values of elastic constants for MgO, MgS, MgSe and MgTe materials fulfilled the Born stability criteria, and these values are also in good agreement with other available results [41-42]. Several critical mechanical parameters can be determined using the elastic constants. Mechanical parameters such as shear modulus (GH), bulk modulus (B0), Young’s modulus (Y), Poisson’s ratio (ν), Pugh’s ratio (B/G), Cauchy pressure (C), and anisotropic ratio (A) are presented in Table 5, respectively. Moreover, in the theory of elasticity, several parameters are used to describe how materials deform under applied forces. Among the most fundamental are the Lame parameters, denoted by λ (lambda) and µ (mu). These parameters are central to the generalized Hookes law for isotropic materials. The parameter µ, also known as the shear modulus or the second Lame parameter, characterizes the materials response to shear stress, that is, how it deforms when forces are applied parallel to its surface. The other Lame parameter, λ, works in tandem with the other to describe volumetric and shear behaviour in materials under stress. Along with these, functional parameters, C generally refers to the elastic stiffness tensor, often written as Cijkl in tensor notation. This tensor governs the relationship between stress and strain in the most general form of Hookes law, especially for anisotropic materials, where different directions in the material respond differently to stress. All these parameters are represented in Table 4.
Another important parameter of a mechanical properties can be determined by using the elastic constants. The mechanical parameters such as shear modulus (GH), bulk modulus (Bo), Young’s modulus (Y), Poisson’s ratio (ν), Pugh’s ratio (B/G), Cauchy’s pressure (C ) and anisotropic ratio (A) which is mentioned in Table 5, respectively. The calculated average Hill’s shear modulus (GH), finding the plastic deformations brought by shear stress. So GH tell us the material’s hardness and equal to arithmetic mean of the Voigt (Gv) and Reuss (GR) moduli. The mathematically expression for GH is [43]:
Where
and
In Table 5, the value of GH reveals that, MgTe3 exist the highest value (great hardness) of GH (23.74 GPa). Young’s modulus tell us the stiffness of a compound [44]. Using the following formula, Young’s modulus may be calculated from the GV and Bo.
MgTe has largest value of Young modulus which is more stiffer than the other materials in Table 5 under study. These materials are harder to break because their Young’s moduli are higher than their bulk and shear moduli. Understanding Pugh’s ratio (B/G) is crucial to comprehending the ductile/brittle character of materials. According to Pugh’s criterion, if B/G > 1.75, the material is considered ductile; if B/G < 1.75, it is considered brittle [45]. For MgTe, B/G = 2.06, which suggests ductile behavior. However, an additional indicator is the Cauchy pressure (C′′ = C12 - C44) [46]. A positive Cauchy pressure typically indicates metallic bonding and ductility, while a negative value suggests directional bonding and brittleness. For MgTe, the Cauchy pressure is negative (-7.85 GPa), which indicates brittle behavior. This apparent contradiction is not uncommon in materials with mixed bonding characteristics. MgTe exhibits a combination of ionic, covalent, and weak metallic bonding. The Pugh ratio is more sensitive to the bulk- to-shear modulus relationship, while Cauchy pressure reflects the angular character of interatomic forces. In MgTe, the directional Mg-Te bonding (covalent/ionic character) likely dominates the shear response, leading to a negative Cauchy pressure. Nevertheless, the relatively low shear modulus compared to the bulk modulus (B/G > 1.75) still allows for some plasticity. Therefore, MgTe is best described as semi-ductile or as a material with mixed mechanical behavior: it may exhibit limited ductility under certain conditions but retains a tendency toward brittleness due to its bonding anisotropy. This interpretation resolves the conflict between the two criteria and is consistent with the calculated Poisson’s ratio (ν = 0.28), which is below 0.33, also suggesting moderate brittleness.
Anisotropic ratio (A) quantifies the intensity of various characteristics in different directions and is given as:
Where A become unity that mean a material is isotropic; otherwise it’s anisotropic nature. Elastic anisotropy is important in the utilization of engineering materials and is closely linked to the likelihood of induced microcracks in a material. Table 5 shows that the anisotropic ratio value is not equal to unity. The departure of A from unity indicates that all of these compounds are elastically anisotropic and that their characteristics vary in various directions. A parameterization of the elastic moduli for homogeneous isotropic material is established by the two parameters λ and µ, which are referred to as Lame’s first and second constants, respectively. Poisson’s ratio and young modulus may be used to compute λ and µ:
and
The value of ν for ductile character is ν >1/3, and for brittle character is ν <1/3 It is clear from Table 5 that MgTe has ν = 0.28, which is below 1/3, further supporting the brittle tendency. Kleinman parameter (ζ) quantifies the internal strain of a material and describes the proportion of bond bending to bond stretching [48]. To obtain the value of ζ, we used the following relation [49, 50]:
The positive value of shear constant Cr(Cr > 0) fulfill the required criteria (Table 5). Hence, all the compounds under study are dynamically stable materials.
Conclusion
The MgTe binary compound was analyzed using DFT, revealing its stable cubic phase with Fm¯3m symmetry and distinct electronic properties. MgTe was found to be a semi- conductor with an indirect band gap of 0.171 eV under the PBE+SOC-GGA approxi- mation. However, using the HSE06 potential, the band gap of MgTe increased to 1.347 eV, which is larger than those obtained with TB-mBJ (1.232 eV) and SCF (0.446 eV). Using the Born criteria, mechanical stability was verified. The mechanical behavior of MgTe shows mixed characteristics: the Pugh ratio (B/G = 2.06) suggests ductility, while the negative Cauchy pressure (-7.85 GPa) indicates brittleness. This contradiction arises from the mixed ionic-covalent bonding nature of MgTe, and the material is best described as semi-ductile. The dynamical and thermal stability of MgTe were confirmed by phonon dispersion curves and AIMD simulations. The results show that MgTe has potential for spintronic and thermoelectric applications due to its narrow indirect band gap and promising ZT values (0.96 for p-type and 0.95 for n-type). In order to fully establish its potential for practical applications in advanced material technologies, additional experi- mental validation is necessary.
Conflicts of Interest
There are no conflicts of interest to declare
Data Availability Statement
The data supporting this study are available within the article and its Supplementary In- formation. Additional datasets, including WIEN2k input files, optimized structures, and raw output data, are available from the corresponding author upon reasonable request. DFT calculations were performed using WIEN2k (version 18.1); phonon dispersion and structural visualization were conducted using PHONOPY and VESTA, respectively. De- tailed computational parameters are described in the Methods section. Sehrish Munsif conceived and supervised the research, while Mazhar Haleem Awan performed the com- putational analysis, interpreted the data, prepared the figures, and acted as corresponding author. Both authors approved the final manuscript.
- Pandey R, Jaffe JE, Kunz AB (1991) Ab initio band-structure calculations for alkaline-earth oxides and sulfides. Physical Review B, 43: 9228.
- Edelstein VM (1990) Spin polarization of conduction electrons induced by electric current in two-dimensional asymmetric electron systems. Solid State Communica- tions, 73: 233-5.
- Amrani B, Benmessabih T, Tahiri M, Chiboub I, Hiadsi S (2006) First principles study of structural, elastic, electronic and optical properties of CuCl, CuBr and CuI compounds under hydrostatic pressure. Physica B: Condensed Matter, 381: 179-186.
- Khenata R, Sahnoun, M, Baltache H, Rrat M, Rached D (2006) Structural, electronic, elastic and high-pressure properties of some alkaline-earth chalcogenides: An ab initio study. Physica B: Condensed Mat- ter, 371: 12-19.
- Singh A, Srivastava V, Aynyas M, Sanyal SP (2009) High pressure struc- tural phase transition and elastic properties of yttrium pnictides. Physica B: Con- densed Matter, 404: 1852-57.
- Garrity EM (2024) Discovering Semiconductors for Power Electronics Through First-Principles Computations. Colorado School of Mines.
- Ghazzy A, Nsairat H, Said R, Sibai OA, AbuRuman A (2024) Magnetic iron oxide-based nanozymes: from synthesis to application. Nanoscale Advances, 6: 1611-42.
- Iqbal R, Bilal M, Jalali-Asadabadi S, Rahnamaye Aliabad HA, Ahmad I (2018) Theoretical investigation of thermoelectric and elastic properties of inter- metallic compounds ScTM (TM= Cu, Ag, Au and Pd). International Journal of Modern Physics B, 32: 1850004.
- Merz WJ (1949) The electric and optical behavior of BaTiO3 single-domain crys- tals. Physical Review, 76: 1221.
- Muller KA, Burkard H (1979) SrTiO3: An intrinsic quantum paraelectric below 4 K. Physical Review B, 19: 3593-02.
- Shirane G, Hoshino S, Suzuki K (1950) X-ray study of the phase transition in lead titanate. Physical Review, 80: 1105.
- Khaliullin G, Maekawa S (2000) Orbital liquid in three-dimensional Mott insulator: LaTiO3. Physical Review Letters, 85: 3950.
- Tokura Y, Nagaosa N (2000) Orbital physics in transition-metal oxides. Sci- ence, 288: 462-8.
- Uti I, Fabian J, Sarma SD (2004) Spintronics: Fundamentals and applica- tions. Reviews of Modern Physics, 76: 323.
- De Groot RA, Mueller FM, van Engen PV, Buschow KHJ (1983) New class of materials: half-metallic ferromagnets. Physical Review Letters, 50: 2024.
- Galanakis I, Dederichs PH, Papanikolaou N (2002) Slater-Pauling behavior and origin of the half-metallicity of the full-Heusler alloys. Physical Review B, 66: 174429.
- Snyder GJ, Toberer ES (2008) Complex thermoelectric materials. Nature Materials, 7: 105-114.
- P Blaha, K Schwarz, F Tran, R Laskowski, G Madsen (2020) “WIEN2k: An APW+lo program for calculating the properties of solids,” 152: 123411123417.
- CJ McCabe, MA Halvorson, KM King, X Cao, DS Kim (2022) “Interpret- ing interaction effects in generalized linear models of nonlinear probabilities and counts,” Multivar. Behav. Res., 57: 243248.
- Z Wu, RE Cohen (2006) “More accurate generalized gradient approximation for solids,” Phys. Rev., 73: 235116.
- P Barlido, T Aull, AW Huran, F Tran, MAL Marques (2019) “Large-scale benchmark of exchange-correlation functionals for the determination of electronic band gaps of solids,” J. Chem. Theory Comput., 15: 50695079.
- T Charpin (2001) “A package for calculating elastic tensors of cubic phase using WIEN,” Laboratoire de Gomtrie, F-75252 Paris, France.
- S Nos (1984) “A unified formulation of the constant temperature molecular dynamics methods,” J. Chem. Phys, 81: 511-519.
- K Momma, F Izumi (2011) “VESTA 3 for three-dimensional visualization of crys- tal, volumetric and morphology data,” J. Appl. Crystallogr., 44: 12721276.
- F Birch (1947) elastic strain of cubic crystals, Phys. Rev., 71: 809.
- M. Born (1940) “On the stability of crystal lattices. I,” in Mathematical Proceedings of the Cambridge Philosophical Society, Cambridge University Press. 36: 160-172.
- Madsen GKH, Singh DJ (2006) “BoltzTraP: A code for calculating band-structure dependent quantities,” Comput. Phys. Commun. 175: 67-71.
- Snyder GJ, Toberer ES (2008) “Complex thermoelectric materials,” Nat. Mater. 7: 105-114.
- Togo A, Tanaka I (2015) “First principles phonon calculations in materials science,” Scr. Mater. 108: 1-5.
- Nose S (1984) “A unifed formulation of the constant temperature molecular dynamics methods”, J. Chem. Phys. 81: 511-19.
- Momma K, Izumi F (2011) “VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data” J. Appl. Crystallogr. 44: 1272-76.
- Kurita K, Koretsune T (2020) Systematic first-principles study of the on-site spin-orbit coupling in crystals. Physical Review B. 102: p.045109.
- Birch F (1947) “Finite elastic strain of cubic crystals” Physical review. 71: 809.
- Khan B, Aliabad HR, Khan I, Jalali-Asadabadi S, Ahmad I (2017) Com- parative study of thermoelectric properties of Co based filled antimonide skutteru- dites with and without SOC effect. Computational Materials Science. 131: 308- 14.
- Mohyedin MZ, Taib MFM, Radzwan A, Shaari A, Mustaffa M, et al. (2020) First principles study of the effect of spin-orbit coupling on thermoelectric properties of Bismuth telluride. Computational and Theoretical Chemistry. 1182: 112851.
- HAMLAT M (2024) “Enhancement of the magneto-electronic properties by GGA and TB-mBJ approaches for KMgO3 perovskite oxide”, Phys. Semicond. Devices & Renew. Energies, 1: 08-08.
- Wang X , Xiaofeng L, Huiqi X, Touwen F, Zhang L, et al. (2023) “Effects of Al and La elements on mechanical properties of CoNiFe0. 6Cr0. 6 high-entropy alloys: a first-principles study”, J. Mater. Res. Technol 23: 1130.
- M Shafiq, I Ahmad, S Jalali Asadabadi (2015) “Mechanical properties and variation in SOC going from La to Nd in intermetallics RIn3 and RSn3 (R = La, Ce, Pr, Nd)”. RSC Advances, 5: 39416.
- Boer TD, Bekheet MF, Gurlo A, Riedel R, Moewes A (2016) “Band gap and electronic structure of cubic, rhombohedral, and orthorhombic In 2 O 3 polymorphs: Experiment and theory” Phys. Rev. B Condens. Matter. 93: 155205.
- Zhi W, Zhao J, Xiang P, Xian H, Meng J (2007) “Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles” Phys. Rev. 5: 76.
- Max B (1940) “On the stability of crystal lattices. I. In: Mathematical proceedings of the Cambridge philosophical society” Cambridge University Press. 36: 160e72.
- AD Becke, MR Roussel (1989) “Exchange holes in inhomogeneous systems: A coordi- natespace model”, Phys. Rev. A, 1989: 3761-67.
- P Barlido, T Aull, AW Huran, F Tran F, MAL Marques (2019) “Large-scale benchmark of exchange-correlation functionals for the detremination of electronic band gaps of solids” J. Chem. Theory Comput., 15: 5069-79.
- R Hill (1952)“The Elastic Behaviour of a Crystalline Aggregate” Proc. Phys. Soc., 1952: 350-4.
- S Qaisi, R Ahmed, BU Haq, S Tahir (2020) “A comprehensive first-principles computational study on the physical properties of lutetium aluminum perovskite LuAlO3”, Mater. Chem. Phys., 250: 123148.
- SF Pugh (1954) “Relations between the elastic moduli and the plastic properties of polycrystalline pure metals” Philos. Mag., 45: 823.
- DG Pettifor (1992) “Theoretical predictions of structure and related properties of inter- metallics”, Mater. Sci. Tech., 1992: 345-49.
- GNAL Greer, RS Lakes, T Rouxel (2011) “Poissons ratio and modern materials” Nature Materials, 10: 823-37.
- L Kleinman (1962) “Deformation potentials in silicon. I. uniaxial strain” Phys. Rev., 1962: 2614-21.
- WA Harrison (1980) “Electronic Structure and the Properties of Solids: The Physics of the Chemical Bond, Freeman” San Francisco.
FIGURE 1
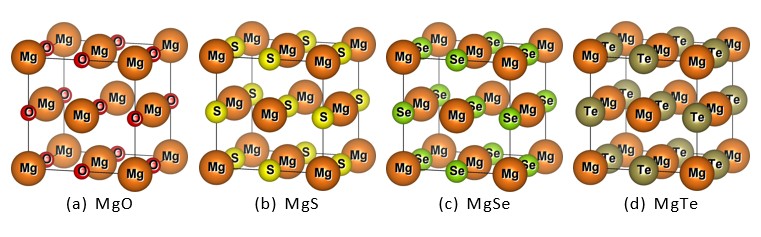
Figure 1: Structural Views of Binary Mgx(X = O, S, Se And Te) Chalcogenides Compounds
FIGURE 2
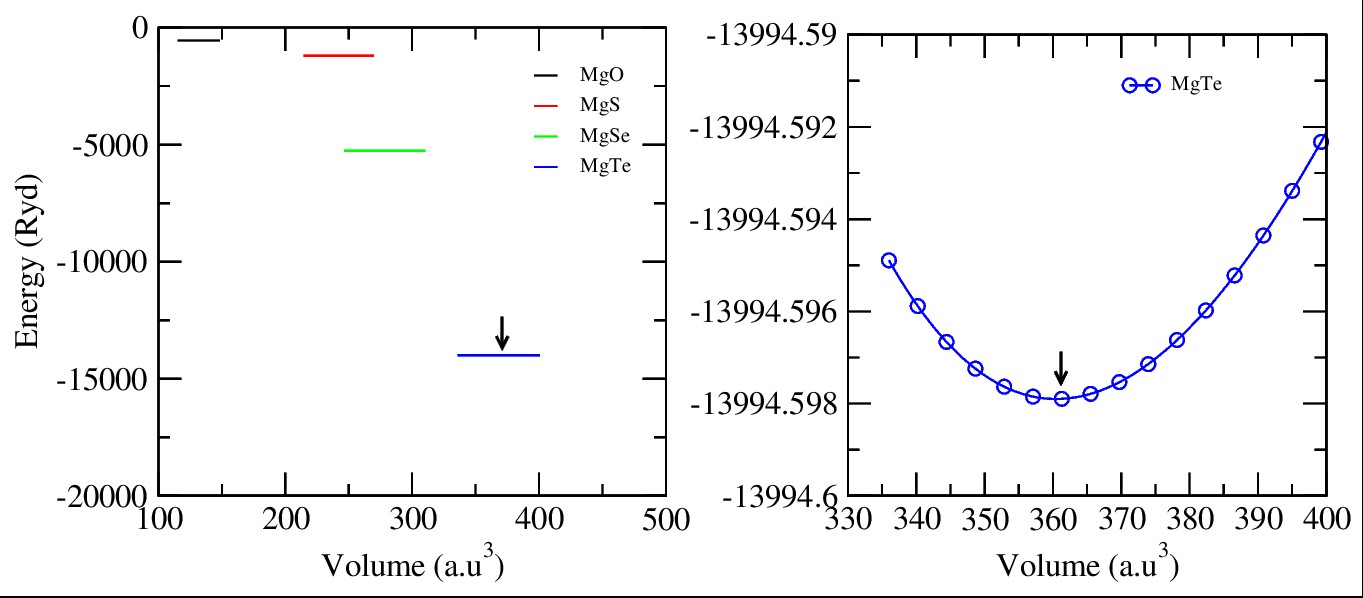
Figure 2: E-V Curves Of Binary Mgx (X = O, S, Se and Te) Compounds Which Correspond to Minimal Energy Point
FIGURE 3
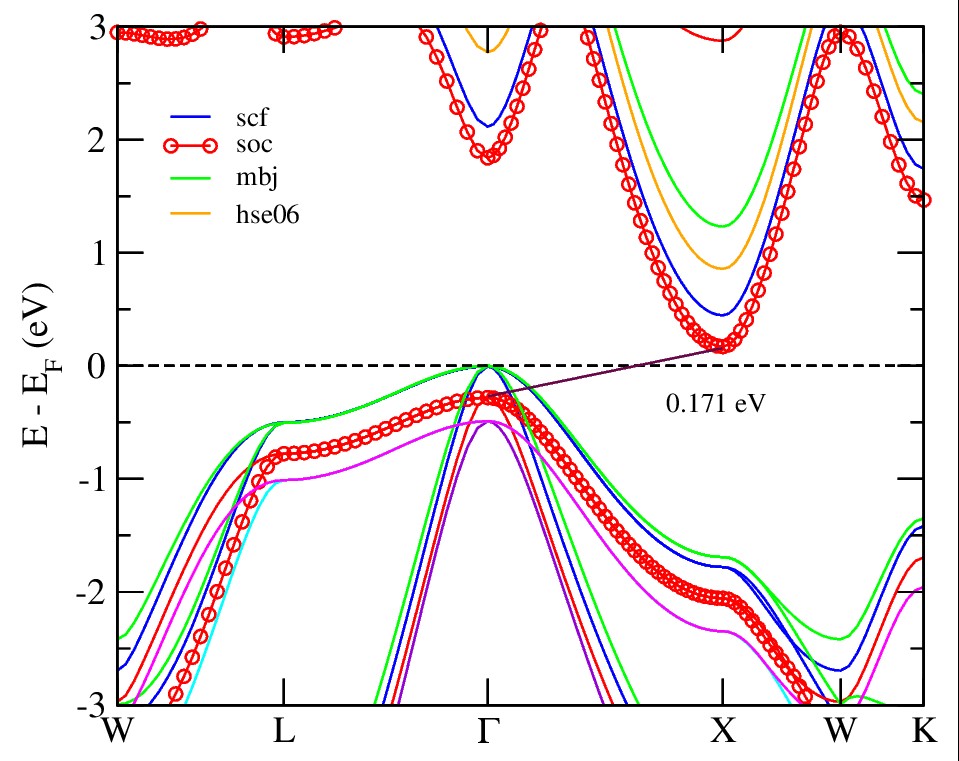
Figure 3: Present the Band Structure of Binary Mgte with Different Potentials
FIGURE 4
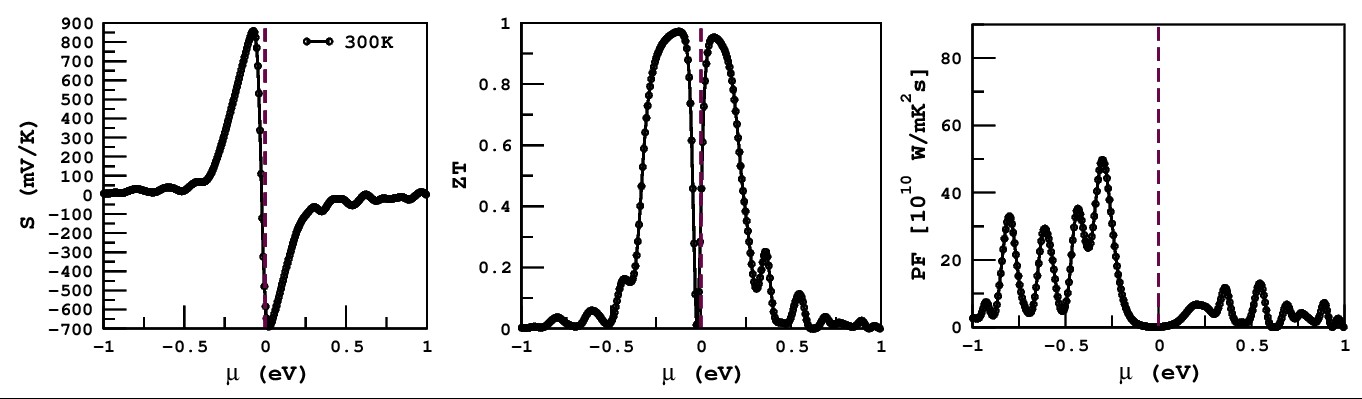
Figure 4: Present the Seebeck, ZT and PF of a binary MgTe with PBE+SOC-GGA
FIGURE 5
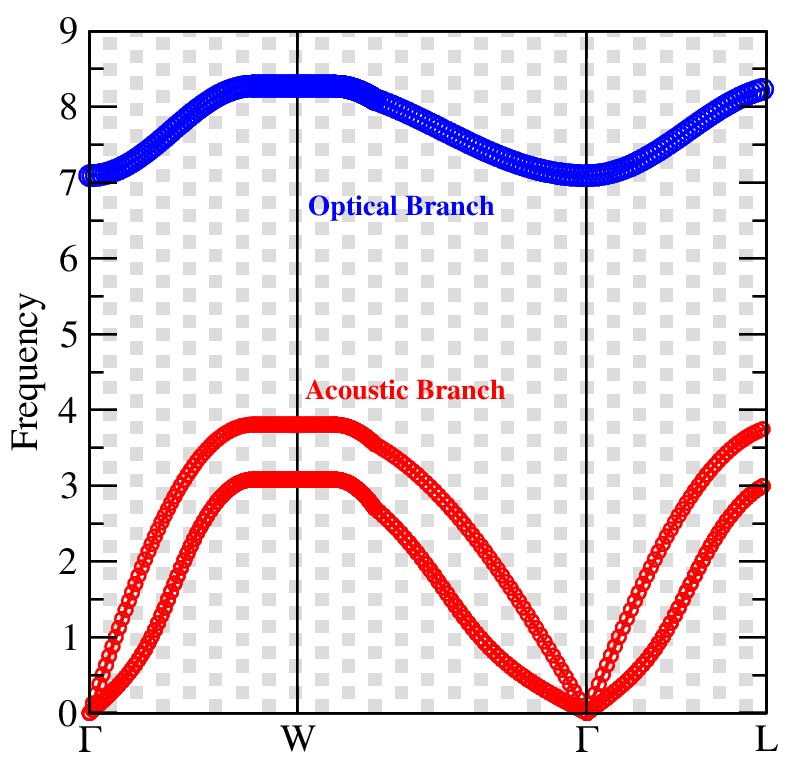
Figure 5: Present the Phononic Dispersion Curve of a Binary Mgte Compound
FIGURE 6
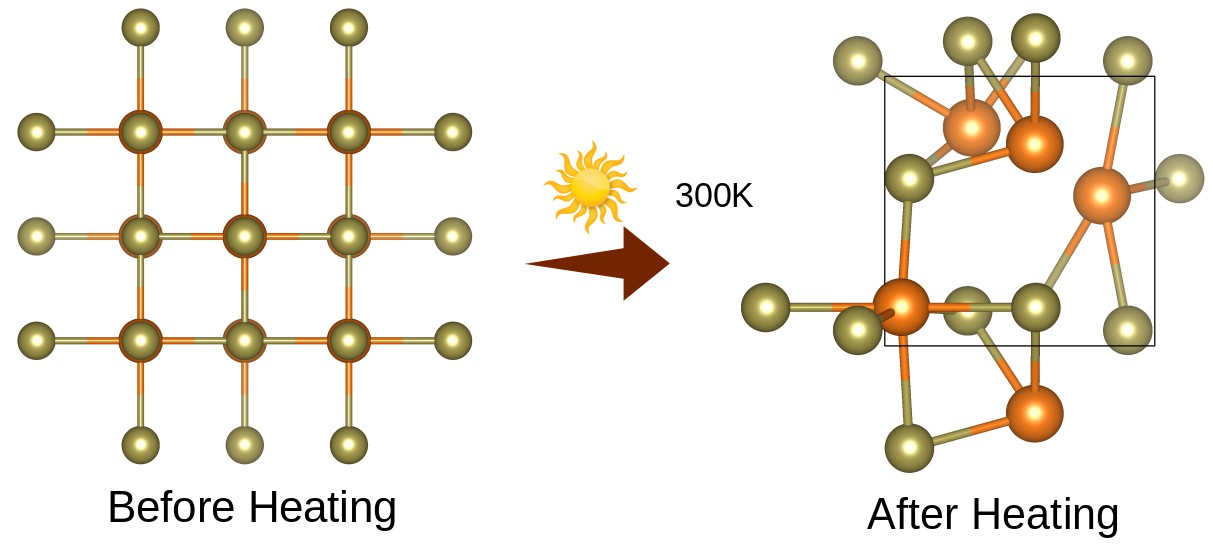
Figure 6: Present the Thermal Stability of a Binary Mgte Before and After Heating during AIMD Simulations
FIGURE 7
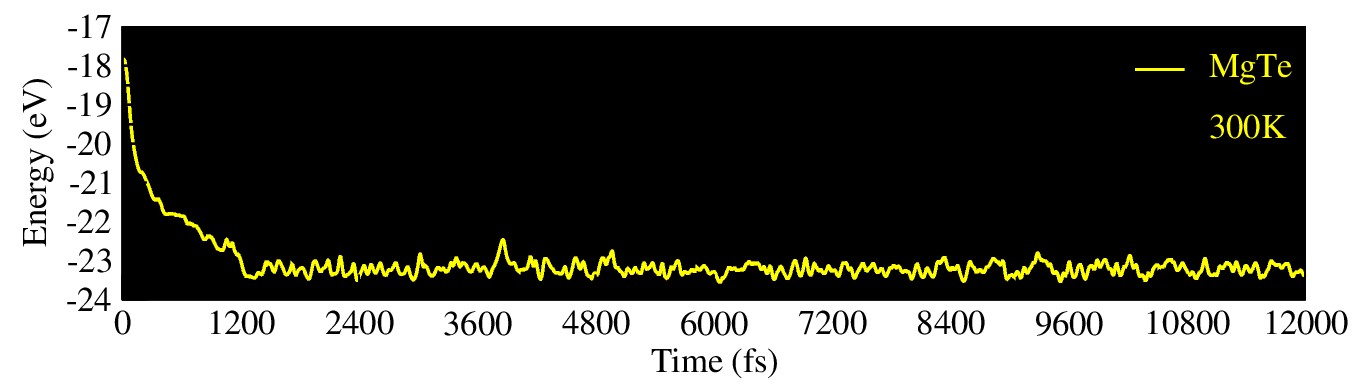
For the phonon cal- culations, MgTe was transformed into a 2 × 1 × 1 supercell with a dense k-mesh of 1 × 1 × 4 to obtain an accurate phonon band structure. Furthermore, the thermal stabil- ity of MgTe was investigated using ab-initio molecular dynamics (AIMD) simulations at 300 K, as illustrated in Figure 7. The simulation was performed for 12 ps with a time step of 1 fs. The AIMD results reveal that the total energy fluctuations remain within a stable range throughout the simulation time, and no structural deformation or bond breaking is observed. Initially, the total energy fluctuates between approximately -17 eV and -23 eV due to thermal excitation; however, after nearly 1200 fs, the energy profile becomes more stable and symmetric, indicating that the system reaches thermal equi- librium. Moreover, no significant distortion is observed before or after heating, which demonstrates that MgTe can effectively withstand room-temperature conditions while maintaining its structural integrity. Overall, these results confirm that MgTe exhibits excellent thermodynamic and thermal stability in its cubic phase at 300 K.
Tables at a glance
Figures at a glance