Epigenetic Modulation of Alzheimer’s Disease Pathology through Methyltransferase Inhibition
Received Date: December 08, 2023 Accepted Date: January 08, 2024 Published Date: January 10, 2024
doi: 10.17303/jnnd.2024.12.101
Citation: Aybuke Seniz Ates, Beyza Yilmaz, Ayyub Ebrahimi (2024) Epigenetic Modulation of Alzheimer’s Disease Pathology through Methyltransferase Inhibition. J Neurophysiol Neurol Disord 12: 1-16
Abstract
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder characterized by the accumulation of amyloid beta 40 and 42 (Aβ40-42) plaques and neurofibrillary tangles in the brain. While Aβ42 is recognized as the most amyloidogenic form due to its hydrophobic structure, AD pathogenesis involves a complex interplay of genetic and epigenetic factors. This study explores the potential therapeutic impact of methyltransferase inhibitors on AD by targeting genes associated with Aβ plaque formation.
To model AD, SH-SY5Y human neuroblastoma cells with a significant presence of Aβ42 were employed. Following cell culture under optimized conditions, the AD model was induced through the Aβ42 protein application. Quantitative assessments of Aβ protein production were conducted using the enzyme-linked immunosorbent assay (ELISA) method. Real Time-Polymerase Chain Reaction (RT-PCR) was employed to measure the expression levels of genes linked to Aβ production, validating the Alzheimer’s model at the genetic level.
Methyltransferase inhibitors were applied to cells at varying concentrations to discern optimal levels, determined through the MTT test. Protein-level quantification of Aβ42 in Alzheimer’s model cells, along with the RT-PCR analysis of genes including APP, PSEN, BACE1, GSAP, and BDNF, revealed promising outcomes. Notably, 5-Azacytidine, 3-Deazaneplanocin, and SGC0946 exhibited a 15%, 17%, and 10% reduction in Aβ42 plaque levels, respectively.
Furthermore, significant alterations in the expression levels of genes associated with Aβ42 plaques were observed, suggesting potential therapeutic benefits. Our findings underscore the essential role of amyloid beta protein in AD and propose that the investigated methyltransferase inhibitors could serve as effective therapeutic tools by mitigating Aβ42 protein levels. This study not only advances our understanding of AD mechanisms but also offers valuable insights into the explora
Keywords: Alzheimer’s Disease; Amyloid beta plaques; Methyltransferase Inhibitors; SGC0946; 3-Deazaneplanocin; 5-Azacytidine
Abbreviations
3-DZNep, 3-Deazaneplanocin A; 5-Aza, Azacytidine; Aβ, amyloid beta; AD, Alzheimer’s disease; APP, amyloid precursor protein; DMEM, Dulbecco’s Modified Eagle Medium; cDNA, Complementary DNA; DMSO, Dimethyl sulfoxide; MTT, 3-4,5-dimethylthiazol-2-yl-2,5-diphenyl-tetrazolium bromide; PS1, human presenilin-1; PSEN2, presenilin-2; qRT-PCR, quantitative real-time PCR; BACE1, beta-site amyloid precursor protein cleaving enzyme 1; ELISA, Enzyme-Linked Immunosorbent Assay; BDNF, Brain-derived neurotrophic factor; GSAP, gamma-secretase activating protein; DNMT, DNA Methyl Transferase; HMT, Histone methyltransferase; KDM, Histone demethylase; KMT, Histone lysine methyltransferases; HDAC, Histone deacetylase; NB, Neuroblastoma; mRNA, messenger RNA; NT, non-treated; PBS, Phosphate-buffered saline; NEAA, Non-Essential Amino Acid; ChIP, chromatin immunoprecipitation.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder marked by cognitive decline, memory loss, and dementia, predominantly affecting individuals over the age of 65 [1,2]. As the leading cause of dementia, accounting for 60-70% of cases, and ranking sixth in mortality within the United States [2], AD poses a significant public health challenge.
Neuropathological changes, including the formation of senile amyloid plaques (SP), neurofibrillary tangles (NFT), and notable brain atrophy, provide crucial insights into the disease’s progression and symptomatic manifestation [3-5]. Amyloid plaques, primarily comprised of amyloid beta (Aβ) peptides, accumulate extracellularly, with Aβ42 being the most amyloidogenic and prevalent form in AD [6-8]. Concurrently, insoluble misfolded tau protein deposits known as neurofibrillary tangles (NFTs) contribute to the neuronal cytoplasmic pathology [9]. The cellular stage, the first stage of AD, occurs in parallel with the accumulation of amyloid β, which induces the spread of tau pathology [10].
While genetic factors, particularly mutations in AD-related genes such as PSEN and APP, are acknowledged contributors to AD risk [11], emerging evidence underscores the pivotal role of epigenetic changes in aging-related diseases, including AD [12]. Notably, DNA methyla tion and histone modifications, governed by enzymes like lysine methyltransferases (KMTs), have been implicated in AD-related gene expression regulation [13-16].
DNA methylation, a fundamental epigenetic mechanism [17], exhibits complex dynamics in AD, with hypermethylation observed in nerve growth factor genes like BDNF and hypomethylation detected in genes like APP, linking to neuropathology [13,18]. In the realm of therapeutic exploration, the last decade has witnessed a focus on promising epigenetic drugs, specifically HDAC inhibitors and histone/DNA-demethylating agents [19].
This study delves into the potential therapeutic effects of methyltransferase inhibitors, specifically Azacytidine, 5-Aza-2’-deoxycytidine (5-Aza), 3-Deazaneplanocin A (3-DZNep), and SGC0946, on AD model SH-SY5Y cells. Azacytidine, inhibiting DNA methyltransferase, induces hypomethylation, leads to decondensation of the chromatin structure, and influences DNA synthesis [20]. 5-Aza- -2’-deoxycytidine (5-Aza) can only be incorporated into DNA because it is a deoxyribonucleoside. With this feature, it is recognized as a substrate by DNA methyltransferases, which prevents DNA synthesis and subsequently leads to cytotoxicity, and covalent binding occurs. As a result, DNA methyltransferase function is blocked. Because of these properties, DNA Methyl Transferase (DNMT) inhibitors such as Azacytidine, which can modulate the methylation of AD risk genes, can change DNA methylation, thereby impairing migration and differentiation [21]. 3-DZNep acts as an inhibitor of S-adenosylhomocysteine hydrolase and a lysine methyltransferase Enhancer of zeste homology 2 (EZH2) which specifically methylate Histone H3 lysine 27 (H3K27) and functions as inactive chromatin mark, showing promise in diseases characterized by active histone methylation, including AD [22-24]. Similarly, SGC0946 exhibits selective inhibition of Disruptor of telomeric silencing-1 like (DOT1L) histone H3K79 methyltransferase which is the active chromatin mark, presenting a novel approach to drug discovery for AD [12,25-26].
Given the alarming projection of dementia cases reaching 130 million by 2050 and the absence of curative treatments for AD, the incorporation of pharmacoepigenetic studies into drug development and personalized therapy becomes imperative [27]. This study aims to comprehensively analyze the molecular effects of DNA and histone methyltransferases in AD model SH-SY5Y cells, shedding light on potential avenues for therapeutic intervention.
Materials and Methods
Cell Culture
The SH-SY5Y human neuroblastoma cell line (ATCC, CRL-2266) was cultured in Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher Scientific, 31885-023) supplemented with 10% FBS (Thermo Fisher Scientific, 16000044), 1% NEAA (Thermo Fisher Scientific, 11140050), and 1% penicillin-streptomycin (Thermo Fisher Scientific, 15140122) in a 37°C incubator with 5% CO2.
Aβ42 Protein Preparation and Application
The Aβ (1-42) Protein Fragment (Sigma-Aldrich, A9810) was provided and prepared as a stock solution in DMSO (Sigma-Aldrich, D2650), following the indicated guidelines and stored at -20°C. SH-SY5Y cells were trypsinized, counted, and seeded at 100,000 cells per well in a 6-well cell culture plate (Corning, 35116). Aβ42 was applied at concentrations of 5 and 10 µM to establish the AD model. After 48 hours of incubation, the media were refreshed. Aβ42-free conditions (non-treated control-NT) served as the negative control. Following an additional 48- hour incubation, media were collected for ELISA analysis and stored at -20°C.
Aβ42 Protein Level Determination
The Aβ42 levels in control and Aβ42-treated cells were measured using the Human Aβ42 ELISA Kit (Invitrogen™, KHB3441) with comparison to the control group, employing the Thermo Scientific™ Multiskan™ GO Microplate Spectrophotometer.
Gene Expression Profiling Assay
Total RNA was extracted using an RNA isolation kit (Zymo Research, R1055). Reverse transcription of 1 μg total RNA to cDNA was performed using a cDNA Synthesis kit (Bioline, BIO-65054) with the Thermal Cycler (Bio-Rad, T100). Quantitative analysis of APP, BACE1, BDNF, GSAP, and PSEN expressions was carried out using a Real-Time PCR Detection System (Bio-Rad, CFX96™ Connect) with Bioline SensiFAST™ SYBR® No-ROX Kit (BIO-98020). mRNA levels were calculated using the 2–∆∆Ct algorithm and normalized to the β-Actin housekeeping gene. Primers were designed using the Primer3 and NCBI Primer Blast online databases (Table 1).
Cell Viability Assay
The 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay was conducted to determine cell viability and optimal chemical concentrations. AD model and control cells were seeded in 96-well plates and treated with varying concentrations of chemicals (5-Aza, 3- DZNep, SGC0946) for 48 hours. 5-Aza is a chemical analogue of the nucleoside cytidine and inhibits DNA methyltransferase, causing hypomethylation of DNA. 3-Deazaneplanocin A (3-DZNep, C-c3Ado) is a chemical that acts as both an inhibitor of S-adenosylhomocysteine hydrolase [22] and a lysine methyltransferase inhibitor. One of the histone methyltransferase inhibitors, SGC0946 inhibits histone H3K79 methyltransferase DOT1L in-vitro by showing strong selective properties for HMTs.
After the AD model and control group cells were seeded in 96-well plates and incubated for 24 hours treated with varying concentrations of chemicals (5-Aza: 1, 5, 10, 50, and 100 µM; 3-DZNep: 0.05, 0,1, 0.5, 1, and 5 µM; SGC0946: 0.1, 0.5, 1, 5, and 10 µM) to determine the optimal dosage and placed in the incubator for 48 hours. 5-Aza (Tocris, 3842) and 3-DZNep (Tocris, 4703) are diluted in H2O, and SGC0946 (Tocris, 4541) is diluted in DMSO. MTT solution was added to each well with a final concentration of 0.5 mg/ml in the medium of the cells and incubated for 3-4 hours at 37ºC. After observing blue crystals, the contents in the wells were spilt out and all wells were washed with PBS.
Then DMSO was added to the wells and incubated for 10 minutes in the dark. After the purple color was observed the plate was measured at 570 nm in the Thermo Scientific™ Multiskan™ GO Microplate Spectrophotometer.
Aβ42 Protein Level Determination and Gene Expression Profiling Assay after Chemical Application in AD-Modelled SH-SY5Y Cells
According to the results obtained after MTT treatment, the Optimal chemical concentrations to be applied to the cells were determined by the lowest concentration that most affected cell viability and cells were then treated in the next step accordingly. After 48 hours, media were collected for ELISA, and cells were harvested for RNA isolation. ELISA and gene expression analysis were performed by comparing protein and gene levels to non-treated (NT) control cells.
According to the results obtained after the MTT application, optimal chemical concentrations were determined according to the lowest concentration that most affected cell viability and cells were treated accordingly.
Statistical Analysis
Three technical and two biological replicates were analyzed for each experiment. Data were interpreted with the significance values of the Multiskan Go microplate reader analysis programs and Bio-Rad CFX ConnectTM RTPCR software, using the Two-tailed Student’s t-test. Statistical significance was considered at p-value ≤ 0.05.
Results
Aβ42 Protein Level Modulation
The impact of varying concentrations of Aβ42 protein on SH-SY5Y cells was assessed through ELISA, revealing a concentration-dependent increase in Aβ42 levels (Fig. 3). Cells treated with 10 µM Aβ42 exhibited a 50% increase in Aβ42 production compared to the control, affirming the successful formation of an Alzheimer’s disease (AD) model.
Aβ42 Treatment Induces the Formation of AD-Model in Gene Expression Level
RT-PCR analysis elucidated alterations in AD-related gene expression levels post-Aβ42 treatment. Notably, APP, BACE1, GSAP, and BDNF gene expressions in creased, while PSEN gene expression decreased in Aβ42- treated cells compared to the control (Figure 4). These findings validate the establishment of the AD model and align with existing literature on AD-related gene expressions.
Methyltransferase Inhibition and Cell Viability
MTT assays determined optimal concentrations for methyltransferase inhibitors (5-Aza, 3-DZNep, SGC0946) on AD model cells. According to the measurements obtained after chemical application, the lowest concentration that reduced cell viability at the highest rate for 5-Aza and 3-DZNep with 14 and 13% decrease was determined as 1 µM (Figure 5a-b). SGC0946 exhibited a concentration-dependent effect, increasing viability at low concentrations and decreasing it at higher concentrations and the optimal lowest concentration which reduced the cell viability at the highest level for SGC0946 (10%) was determined as 5 µM (Figure 5c).
Methyltransferase Inhibition and Aβ42 Protein Levels
Following optimal concentration determination, methyltransferase inhibitors reduced Aβ42 levels in AD-- modelled cells. Moreover, sustained Aβ42 protein production was observed after multiple passages of Aβ42-treated cells, underscoring the chronic nature of Aβ42 induction (Figure 6).
Methyltransferase Inhibition and Gene Expression Levels
qRT-PCR analysis post-chemical application revealed that 5-Aza, 3-DZNep, and SGC0946 influenced the expression of AD-related genes. Notably, APP, GSAP, and PSEN expressions were modulated, affirming the potential of these methyltransferase inhibitors in regulating gene expressions associated with AD pathology (Figure 7a-c).
Discussion
Genetic, epigenetic and environmental factors play a role in the emergence of AD [28]. Among the genetic factors, autosomal dominant inherited mutations in APP, PSEN1 and PSEN2 take the first place [29]. The fibrils formed by Aβ, form plaques in the brains of AD patients. Aβ is produced from amyloid precursor protein (APP) by sequential proteolytic cleavage involving β-secretase (βeta-site APP cleaving enzyme1) and γ-secretase [30]. The gene encoding the γ-Secretase activating protein (GSAP) plays an important role in the regulation of γ-secretase activity and specificity [31]. One of the factors required to produce Aβ42, which initiates toxicity in AD, and all other monomeric forms of Aβ, is the Beta-beta site APPamyloid precursor protein cleaving enzyme 11(BACE1). Therefore, BACE1 activity rates increase in AD patients [32]. Accordingly, BACE1 can be used as a biomarker for the early diagnosis of Alzheimer’s disease and can be a targeted protein to stop Aβ accumulation. BDNF is a growth factor that acts on β cells and has an important role in neuronal development in the brain. Brain BDNF levels are low in patients with neurodegenerative diseases such as Alzheimer’s or Parkinson’s disease [33].
One of the most effective mechanisms in AD is epigenetic modifications, which are the changes that do not occur in the DNA sequence but change the expression of genes. The most common epigenetic changes in Alzheimer’s disease are DNA/histone methylations. Based on this information, it is thought that therapeutics targeting these modifications will provide good results in AD [34].
According to the amyloid hypothesis, Aβ accumulation in the brain is the main factor driving the pathogenesis of AD [35]. Based on this hypothesis, we aimed to create an AD model in cells by triggering amyloid beta production by applying the Aβ42 protein to SH-SY5Y cells. Studies show that the appropriate concentration of Aβ42 protein administered when creating an AD model in SH-SY5Y cells is between 5-15 μM [51,52,53]. After 5 and 10 µM Aβ42 protein application separately, cells were exposed to Aβ42 for 48 hours and replaced with fresh media that did not contain Aβ42. After two more days of incubation, as a result of ELISA measurements, it was determined that the cells treated with 10 µM Aβ42 had a much higher concentration of Aβ42 protein release than control cells, compared to cells treated with 5 µM Aβ42 protein.
According to the results obtained by qRT-PCR for the quantitative measurement of gene expression in AD-- modelled cells, an increase was observed in the expression of the APP gene, while a decrease was detected in the expression of the PSEN gene. Studies have shown that BACE1 protein is elevated in brain regions affected by AD [36,37] and in line with these data the high increase in BACE1 expression level (Fig. 4) we obtained in our study, showed that Aβ42 administration had a significant effect on BACE1 in cells. A significant increase in GSAP gene expression was detected as a result of qRT-PCR performed on the cells we created the AD model (Fig. 4), and this result shows that data compatible with the literature were obtained and clearly explains the increase in amyloid levels in the cells. As a result of the qRT-PCR analysis we performed, it was determined that the BDNF expression level in AD-modelled SH-SY5Y cell lines increased (Fig. 4). This increase suggested that, since BDNF is a protein that supports the survival of nerve cells, it may be their first reaction against the toxicity of pathological Aβ42 applied to the cells.
Studies in the field of epigenetics have shown that histone modifiers are highly associated with pathological features of AD, such as abnormal tau phosphorylation and Aβ protein plaques [38]. In regulating some of the histone methylation levels that occur with age, HMTs and KDMs (Histone lysine demethylases) are crucial in maintaining appropriate levels of histone methylation marks. A proper balance between histone methylation modifying enzymes is essential because an increase or decrease in the levels and activity of these enzymes and the consequent changes in the methylation levels can contribute to disease states such as AD [39]. Based on this information, the cell viability assay was performed to determine the optimum concentration for chemicals that will reduce the Aβ42 level in SH-SY5Y cells, for which we created AD-modelled cells, and to analyze the viability of the cells exposed to this concentration. Our cell viability assay results showed that 5-Aza which is a DNMT inhibitor, reduces the cell proliferation of the AD-- modelled SH-SY5Y cell lines by 10-15% in all concentrations. Another study analyzed the methylation levels of CpG islands in enhancer regions in AD, showing that epigenetic dysregulation in these regions is associated with AD pathology and may affect neuronal health [40]. This reduction in the activity of methyltransferases may also affect lots of the genes in the cells. A related study showed that the expression of the APP gene was increased in the brain of an AD patient with hypomethylation [41]. Another study showed that the expression of genes such as PSEN1 and BACE1 can be regulated by methylation [42].
There are not many studies to determine the role of histone methylation in the pathogenesis of AD. Studies have shown that high expression of EZH2 is correlated with the poor prognosis of neuroblastoma (NB) patients [43]. At the same time, a study on AD and aging showed that the expression of many genes, including NRF1, an activator of genes regulating cellular growth, and REST, known as the neuron-restricting factor, is regulated by EZH2 [44]. Our results showed that 3-DZNep, an EZH2 inhibitor, at different concentrations significantly reduced cell viability in both control SH-SY5Y neuroblastoma cells and Aβ42 treated cells, as expected. This suggests that inhibition of EZH2 histone methyltransferase may affect the expression of genes involved in cell viability and senescence in AD cells.
Another chemical we used in our study, SGC0946, is an inhibitor of DOT1L. All applied concentrations of SGC0946 increased viability in control cells. However, as the applied concentration increased, the viability rate increased in control cells began to decrease, and a significant decrease in cell viability was observed in Aβ42 cells as expected. When choosing the appropriate concentration for SGC0946, we paid attention to the concentration that had the least effect on the negative control cells, because increasing viability would be as undesirable as decreasing it, since the cells we examined were NB cell lines. It was thought that due to the fact that an epigenetic modification that increases the expression of genes that suppress proliferation in cells, silencing these genes with an epigenetic suppressor such as SGC0946, activates the proliferation of cells. However, it was observed that the cells could not resist high doses and began to decrease viability in Aβ42 cells compared to control cells.
After determining the optimum concentrations of the chemicals to be used, ELISA analysis for the measurement of Aβ42 level after non-treated control and Alzheimer’s model cells, which were applied chemicals at determined concentrations, revealed that 3-DZNep, SGC0946 and 5-Aza reduced the Aβ42 level at a high rate. In addition, when the first ELISA analysis for the measurement of Aβ42 level was compared when the AD-modelled cells were created in the cells, and the ELISA analysis performed on Aβ42 cells, which were advanced 3-4 passages for chemical application, it was seen that these cells started to produce Aβ42 at a much higher rate than control cells. After observing these reducing effects of the epigenetic inhibitors we applied on AD-modelled cells on Aβ42 levels, we thought that these drugs might have a similar effect at the gene level.
As a result of qRT-PCR analysis performed for the analysis of the effects of epigenetic inhibitors applied to cells at the gene level it was observed that the expression of APP, GSAP which is a γ-secretase activator, and PSEN, functioning as a subunit of γ-secretase involved in the degradation of APP decreased by the application of 5-Aza (1 µM) in the AD-modelled cells (Fig. 7). These results support that 5- Aza lowers Aβ42 level in AD cells at gene level compared to control, as we observed in our data obtained by ELISA (Fig. 6). This suggests that 5-Aza can reduce early Aβ42 accumulation in AD by reducing the expression of APP, PSEN and GSAP genes encoding the proteins that lead to Aβ42 formation. We have observed that 3-DZNep (1 µM) and SGC0946 (5 µM) in AD-modelled cells lead to a decrease in GSAP and an increase in BDNF gene expression (Fig. 7). Similar to the results we obtained from 5-Aza, we think that 3- DZNep and SGC0946 HMT inhibitors may also decrease the intracellular Aβ42 peptide level by reducing GSAP gene expression. In conclusion, our qRT-PCR results support at the gene level that 5-Aza, as well as 3-DZNep and SGC0946 HMT inhibitors, lower the Aβ42 level in AD cells compared to the control, as we observed in our ELISA data (Fig. 6). In addition, the increase in BDNF gene expression suggests that 3-DZNep and SGC0946 will contribute to the protection of neuronal cells in a disease characterized by a high level of neurodegeneration, which is also associated with low BDNF.
Based on the knowledge that DNA methylation dynamics are key components of epigenetic regulation in the mammalian central nervous system [45], we wanted to target DNA and histone methylation in this study. 5-Azacitidine was discovered many years ago and has been shown to be incorporated into DNA and inhibit DNA methylation [46]. 3-DZNep, an HMT inhibitor that we applied in our study, is a chemical that causes the degradation of the EZH2 complex at the protein level. EZH2 is a methyl transferase enzyme involved in H3K27 methylation. The idea of therapeutically targeting this enzyme, which is sensitive to the inhibition of 3-DZNep, arose when its overexpression was detected in various cancer diseases [47]. The results we obtained at the end of the application supported this data. DOT1L is one of the HMTs involved in the methylation of histone H3 at the Lysine 79 (H3K79) position [48]. Studies have shown that this methylation mark increases with aging. Due to the ability of DOT1L to change the level of gene expression, strategies to inhibit DOT1L were thought to be a rejuvenation-based approach [25]. Based on this, we used SGC0946 as a DOT1L inhibitor in this study. The results showed that it positively altered the expression of AD-related genes in Aβ42 cells treated with 5 µM SGC0946.
The results underscore the pivotal role of Aβ42 in AD pathogenesis, with Aβ42 protein levels correlating with altered gene expressions. Methyltransferase inhibitors demonstrated significant impacts on cell viability, Aβ42 levels, and gene expressions, indicating their potential as therapeutic tools in mitigating AD-related pathologies.
Conclusions and Future Perspectives
This study illuminates the complex interplay between Aβ42, gene expressions, and methyltransferase activity in AD. The observed effects of methyltransferase inhibitors on cell viability, Aβ42 levels, and gene expressions open avenues for targeted therapeutic interventions. Future investigations, including RNA and ChIP-sequencing, are warranted to comprehensively understand epigenetic factors in AD treatment. Expanding the repertoire of chemicals and using diverse AD cell models will contribute to uncovering intricate mechanisms and developing effective treatments for this challenging disease.
Acknowledgement
This study was supported by the Molecular Biology and Genetics Department at Haliç University (Turkey).
- Godyń J, Jończyk J, Panek D, Malawska B (2016) Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep 68: 127-38.
- Kumar A, Sidhu J, Goyal A, Tsao JW (2022) Alzheimer Disease. In: StatPearls. Treasure Island (FL): StatPearls Publishing.
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer’s disease. Cold Spring Harb Perspect Med 1: a006189.
- Spires-Jones TL, Hyman BT (2014) The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82: 756-71.
- Singh SK, Srivastav S, Yadav AK, Srikrishna S, Perry G (2016) Overview of Alzheimer’s Disease and Some Therapeutic Approaches Targeting Aβ by Using Several Synthetic and Herbal Compounds. Oxid Med Cell Longev 7361613.
- Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G (1991) Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci U S A 88: 7552-6.
- Purves D, Augustine GJ, Fitzpatrick D, Hall WC, LaManita AS, White LE et al. (2012) Neuroscience (5th ed.). Sunderland, MA: Sinauer Associates 713.
- Walker LC (2020) Aβ Plaques. Free Neuropathol. 1:31.
- Sontheimer H (2015) Diseases of the Nervous System. Chapter 4 - Aging, Dementia, and Alzheimer Disease 99-131.
- Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G et al. (2021) Alzheimer’s disease. Lancet (London, England), 397: 1577-90.
- Khanahmadi M, Farhud DD, Malmir M (2015) Genetic of Alzheimer’s Disease: A Narrative Review Article. Iranian journal of public health 44: 892–901.
- Cacabelos R, Torrellas C (2015) Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response. Int J Mol Sci 16: 30483-543.
- Esposito M, Sherr GL (2019) Epigenetic Modifications in Alzheimer’s Neuropathology and Therapeutics. Front Neurosci 13: 476.
- Miller CA, Sweatt JD (2008) Covalent modification of DNA regulates memory formation published correction appears in Neuron 59: 1051. Neuron 53: 857-69.
- Day JJ, Sweatt JD (2010) DNA methylation and memory formation. Nat Neurosci 13: 1319-23.
- Al Temimi, AHK, Amatdjais-Groenen HIV, Reddy YV et al. (2019) The nucleophilic amino group of lysine is central for histone lysine methyltransferase catalysis. Commun Chem 2: 112.
- Anderson OS, Sant KE, Dolinoy DC. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J Nutr Biochem 23: 853-9.
- Gasparoni G, Bultmann S, Lutsik P, et al. (2018) DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 11: 41.
- Bufill E, Ribosa-Nogué R, Blesa R (2020) The Therapeutic Potential of Epigenetic Modifications in Alzheimer’s Disease. In X. Huang (Ed.), Alzheimer’s Disease: Drug Discovery. Exon Publications.
- Müller A, Florek M (2010) 5-Azacytidine/Azacitidine. In: Martens, U. (eds) Small Molecules in Oncology. Recent Results in Cancer Research 184.
- Singh RP, Shiue K, Schomberg D, Zhou FC (2009) Cellular epigenetic modifications of neural stem cell differentiation. Cell Transplant 18: 1197-211.
- Fiskus W, Wang Y, Sreekumar A, et al. (2009) Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 114: 2733-43.
- Gray SG (2012) The Potential of Epigenetic Compounds in Treating Diabetes, In Translational Epigenetics, Epigenetics in Human Disease, Chapter 17: 331-67.
- Miranda TB, Cortez CC, Yoo CB, et al. (2009) DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther 8: 1579-88.
- Soria-Valles C, Osorio FG, López-Otín C (2015) Reprogramming aging through DOT1L inhibition. Cell Cycle 14: 3345-6.
- Yu W, Chory EJ, Wernimont AK, et al. (2013) Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors published correction appears in Nat Commun. 2013;4:1893.. Nat Commun 3: 1288.
- Dubois B, Feldman HH, Jacova C, et al. (2014) Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria published correction appears in Lancet Neurol 13: 757. Lancet Neurol 13: 614-29.
- Alagiakrishnan K, Gill SS, Fagarasanu A (2012) Genetics and epigenetics of Alzheimer’s disease. Postgrad Med J 88: 522-9.
- Sheppard O, Coleman M (2020) Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. In: Huang X, ed. Alzheimer’s Disease: Drug Discovery. Brisbane (AU): Exon Publications.
- Hur JY (2022) γ-Secretase in Alzheimer’s disease. Exp Mol Med 54: 433-46.
- Xu P, Chang JC, Zhou X, et al. (2021) GSAP regulates lipid homeostasis and mitochondrial function associated with Alzheimer’s disease. J Exp Med 218: e20202446.
- Hampel H, Vassar R, De Strooper B, Hardy J, Willem M et al. (2021) The β-Secretase BACE1 in Alzheimer’s Disease. Biological psychiatry 89: 745-56.
- Bathina S, Das UN (2015) Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci 11: 1164-78.
- Sharma VK, Mehta V, Singh TG (2020) Alzheimer’s Disorder: Epigenetic Connection and Associated Risk Factors. Curr Neuropharmacol 18: 740-53.
- Prasansuklab A, Tencomnao T (2013) Amyloidosis in Alzheimer’s Disease: The Toxicity of Amyloid Beta (Aβ), Mechanisms of Its Accumulation and Implications of Medicinal Plants for Therapy. Evid Based Complement Alternat Med. 413808.
- Yang LB, Lindholm K, Yan R, et al. (2003) Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer’s disease. Nat Med 9: 3-4.
- Johnston JA, Liu WW, Todd SA, et al. (2005) Expression and activity of beta-site amyloid precursor protein cleaving enzyme in Alzheimer’s disease. Biochem Soc Trans 33: 1096-100.
- Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M (2015) Increased acetyl and total histone levels in post-- mortem Alzheimer’s disease brain. Neurobiol Dis 74: 281-94.
- Santana DA, Smith MAC, Chen ES (2023) Histone Modifications in Alzheimer’s Disease. Genes (Basel) 14: 347.
- Li P, Marshall L, Oh G, Jakubowski JL, Groot D et al. (2019) Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nature communications, 10: 2246.
- West RL, Lee JM, Maroun LE (1995) Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. Journal of molecular neuroscience: MN 6: 141-6.
- Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S (2005) S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Molecular and cellular neurosciences, 28: 195-204.
- Li Z, Takenobu H, Setyawati AN, et al. (2018) EZH2 regulates neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications. Oncogene 37: 2714-27.
- Ciryam P, Kundra R, Freer R, Morimoto RI, Dobson CM, Vendruscolo M (2016) A transcriptional signature of Alzheimer’s disease is associated with a metastable subproteome at risk for aggregation. Proceedings of the National Academy of Sciences of the United States of America, 113: 4753-8.
- Irier HA, Jin P (2012) Dynamics of DNA methylation in aging and Alzheimer’s disease. DNA Cell Biol 31: S42-8.
- Christman JK (2002) 5-Azacytidine and 5-aza- -2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21: 5483-95.
- Pande V, Pocalyko DJ, Dhanak D (2015) Emerging Epigenetic Therapies—Lysine Methyltransferase/PRC Complex Inhibitors. Epigenetic Cancer Therapy 427-37.
- Bovio PP, Franz H, Heidrich S, et al. Differential Methylation of H3K79 Reveals DOT1L Target Genes and Function in the Cerebellum In Vivo. Mol Neurobiol. 2019;56(6):4273-4287. doi:10.1007/s12035-018-1377-1.
- Tcw J, Goate AM (2017) Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb Perspect Med 7: a024539.
- Shen J, Kelleher RJ (2007) 3rd. The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A 104: 403-9.
- Zatsepina OG, Kechko OI, Mitkevich VA, et al. (2018) Amyloid-β with isomerized Asp7 cytotoxicity is coupled to protein phosphorylation. Sci Rep 8: 3518.
- Mitkevich VA, Petrushanko IY, Yegorov YE, et al. (2013) Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells. Cell Death Dis. 4: e939.
- Yurinskaya MM, Mitkevich VA, Kozin SA, Evgen’ev MB, Makarov AA, Vinokurov MG (2015) HSP70 protects human neuroblastoma cells from apoptosis and oxidative stress induced by amyloid peptide isoAsp7-Aβ(1-42). Cell Death Dis 6: e1977.
FIGURE 1
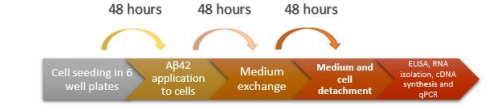
Figure 1: Stages of AD Model Forming. The timetable of different stages of creating the AD model with SH-SY5Y cell lines
FIGURE 2
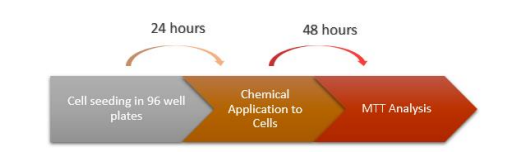
Figure 2: Chemical Application Stages to SH-SY5Y Cells. The timetable for application of different inhibitors of methyltransferases in the AD model cell lines
FIGURE 3
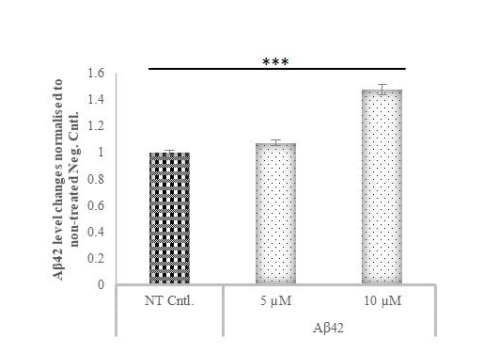
Figure 3: Aβ42 levels in cell media after treatment with two different concentrations of Aβ42 compared to the control group
Aβ42 levels in media after treatment with Aβ42 protein at 5 and 10 μM compared with the control groups by ELISA. Cells treated with 10 μM
Aβ42 produced more Aβ42 than cells treated with 5 μM. All results were determined according to the kit standards obtained by measurement
at 450 nm and by calculating the regression value (R2
=0.8394). *** p≤value = 0.001.
FIGURE 4
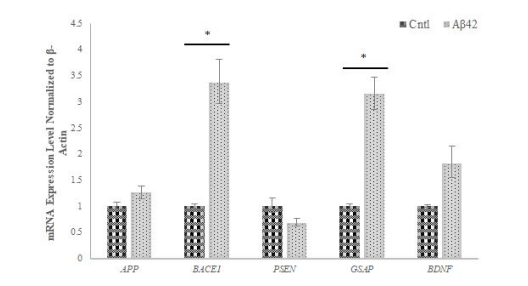
Figure 4: Gene expression profiling of AD-related selected genes
It was observed that the expression of APP, BACE1, GSAP and BDNF genes increased in Aβ42 cells compared to the control, while the expression of the PSEN gene decreased compared to the control. The data represent the mean mRNA expression level of selected genes normalized to β-Actin ± Standard Deviation (SD) from three independent experiments performed in duplicate. * p≤value = 0.05.
FIGURE 5
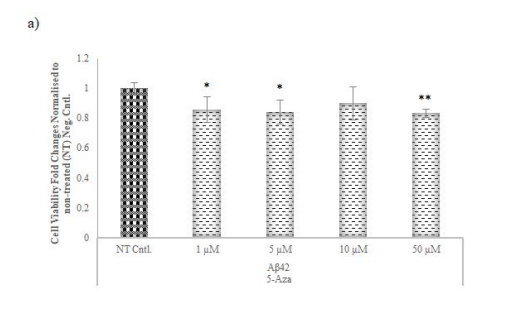
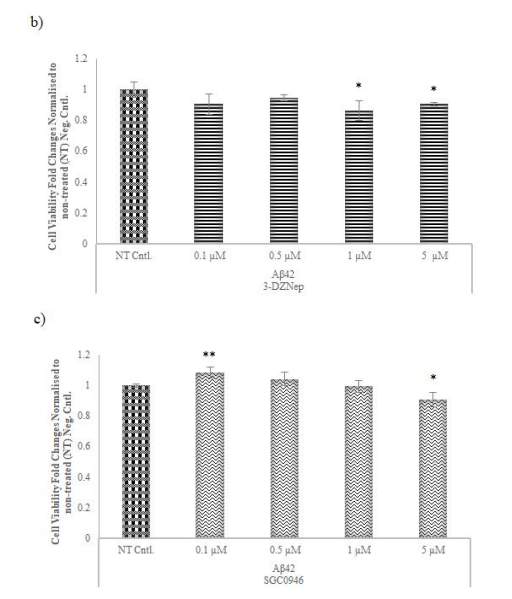
Figure 5: Cell viability assay for AD-modelled cell lines with different concentrations of the chemicals used to inhibit the selected methyltransferases. To examine the optimal dosage of chemicals used to inhibit the activity of a) 5-Azacytidine, b) 3-Deazanoplanocin, c) and SGC0946 of methyltransferases MTT assay performed after 48 hours in AD-modelled SH-SY5Y cell lines
Cell viability in Aβ42 cells treated with 5-Aza and 3-DZNep was reduced relative to NT at each concentration. An increase in cell viability was observed in Aβ42 cells treated with SGC0946 compared to NT cells at minimum concentration, and a decrease in cell viability was observed depending on the high concentrations. * p≤value = 0.05 and ** p≤value = 0.01.
FIGURE 6
Figure 6: Aβ42 Protein level changes after chemical application in AD-modelled cell lines
Aβ42 Protein levels in media after treatment with 10 μM of Aβ42 protein and chemicals used to inhibit the activity selected methyltransferases compared with the non-treated SH-SY5Y negative control by ELISA. Aβ42 level increased in Aβ42 NT cells compared to SH-SY5Y negative control. Histone and DNA methyltransferase inhibitors applied to AD-modelled cells reduced Aβ42 levels by 15%, 17%, and 10% in 5- Azacytidine, 3-Deazaneplanocin, and SGC0946 treated cells compared to NT control cells, respectively. All results were determined according to the kit manual and standards obtained by measurement at 450 nm and by calculating the regression value (R2 =0.99). ** p≤value = 0.01.
FIGURE 7
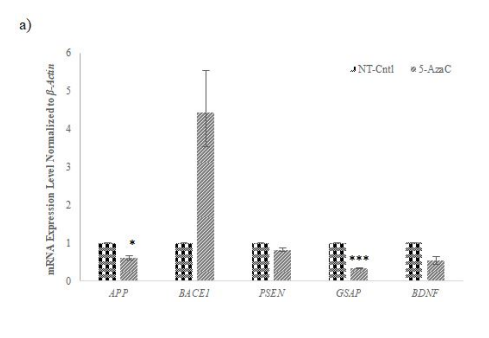
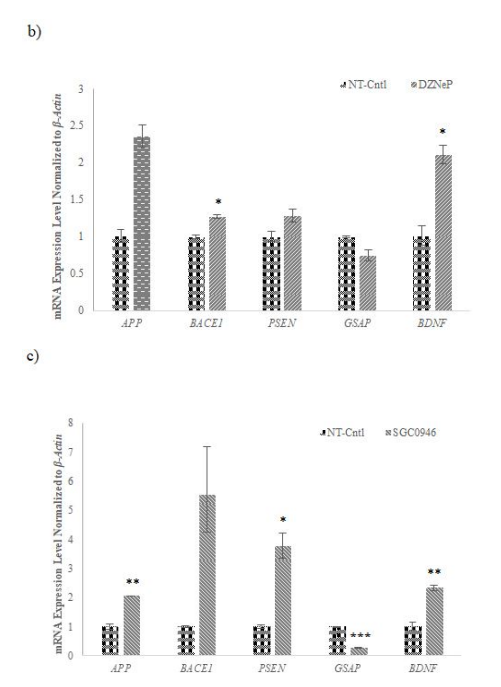
Figure 7: Gene expression profiling of AD-related selected genes in AD-modelled cell lines treated with different chemicals used to inhibit the selected methyltransferases
mRNA expression levels of selected AD-related genes normalized to housekeeping gene expression level performed by qRT-PCR for a) 5-Azacytidine (5-Aza, 1 μM), b) 3-Deazanoplanocin (3-DZNep, 1 μM), c) and SGC0946 (5 μM) of methyltransferases after 48 hours. While the expression of BACE1 increased with 5-Aza application, the expression of other genes decreased compared to NT cells. While the expression of the GSAP gene decreased in cells treated with 3-DZNep and SGC0946, the expression of other genes increased compared to NT cells. The data represent the mean mRNA expression level of selected genes normalized to β-Actin ± SD. from three independent experiments performed in duplicate. * p≤value = 0.05, ** p≤value = 0.01, *** p≤value = 0.001.
Tables at a glance
Figures at a glance