Phase I Study of Lurbinectedin in Japanese Patients with Pretreated Advanced Solid Tumors
Received Date: May 28, 2023 Accepted Date: June 28, 2023 Published Date: July 01, 2023
doi: 10.17303/jocr.2023.4.106
Citation: Shunji Takahashi, Toshihiko Doi, Jose Antonio Lopez-Vilarino, Carmen Kahatt, Mariano Siguero et al. (2023) Phase I Study of Lurbinectedin in Japanese Patients with Pretreated Advanced Solid Tumors. JJ Oncol Clin Res 4: 1-19
Abstract
Background: The recommended dose (RD) of lurbinectedin in non-Japanese patients is 3.2 mg/m2 on Day 1 every three weeks (q3wk).
Methodology: In this phase I trial, Japanese patients with unresectable/advanced solid tumors were treated at 1.5, 2.5 and 3.2mg/m2 q3wk without primary granulocyte colony-stimulating factor (G-CSF) prophylaxis, or at 3.2 and 3.5 mg/m2 q3wk with primary G-CSF prophylaxis, using a classical 3+3 escalation design to determine the RD and suitable schedule.
Results: Initial dose escalation in 15 Japanese patients treated with lurbinectedin without primary G-CSF prophylaxis resulted in a RD of 2.5 mg/m2. Grade ≥3 neutropenia was the main dose-limiting toxicity (DLT) observed in two of four patients treated at the maximum tolerated dose (MTD, 3.2 mg/m2). Treatment-related AEs were mild/moderate. A new dose escalation with primary G-CSF prophylaxis was done in 11 Japanese patients. At the RD defined for lurbinectedin with primary G-CSF prophylaxis (3.2 mg/m2), two of nine patients had DLTs (grade 4 thrombocytopenia), leading to dose reduction. The most common grade ≥3 toxicities at the RD were neutropenia (22%; grade 4, 11%), grade 4 thrombocytopenia, and grade 3 anemia (22% each). Myelotoxicity was reversible and manageable, and non-hematological toxicities were mild/moderate.The pharmacokinetic profile of lurbinectedin was similar to that observed in non-Japanese patients.
Conclusion: Lurbinectedin at 3.2 mg/m2 with primary G-CSF prophylaxis showed an acceptable, predictable and manageable safety profile with hints of antitumor activity in Japanese patients with unresectable and advanced solid tumors, and is the recommended schedule for phase II studies.
Keywords: Lurbinectedin; Pharmacokinetics; Japanese Patients; Phase I Study; Solid Tumors
Introduction
Lurbinectedin (Zepzelca®) is a synthetic tetrahydroisoquinoline alkaloid structurally related to trabectedin.Lurbinectedin inhibits oncogenic transcription primarily through binding to the exocyclic amino group of guanine-rich DNA sequences around the promoters of protein--coding genes, thereby altering the 3D DNA structure and evicting oncogenic transcription factors from their binding sites, halting their aberrant transcription programs [1-3].Lurbinectedin adducts can also stop transcribing (phosphorylated) RNA polymerase II, decreasing mRNA synthesis,and inducing the ubiquitination and degradation of RNA polymerase II inhibition [4]. Lurbinectedin adducts may also trick the nucleotide excision repair system, favoring the production of DNA double-strand breaks and triggering apoptotic cell death [5].
The recommended dose (RD) for lurbinectedin administered as a 1-hour intravenous (i.v.) infusion on Day (D)1 every 3 weeks (q3wk) in non-Japanese patients with cancer in studies conducted in the US and Europe was 3.2mg/m2 [6-8]. In a Basket phase II study, nine cohorts of patients with different difficult-to-treat tumor types received lurbinectedin 3.2 mg/m2 to establish the proof-of-concept of anticancer activity for further clinical development.Based on the results in the small cell lung cancer (SCLC) cohort [7], approval of lurbinectedin was first obtained in the US [9] and later in other countries (Canada, Australia, Singapore, Arab Emirates and Qatar). Lurbinectedin is recommended in the United States National Comprehensive Cancer Network (NCCN) guidelines [10] and the European Society of Medical Oncology (ESMO) [11] as a treatment option for relapsed SCLC. Recently, the results of another cohort of this Basket trial showed relevant antitumor activity in relapsed Ewing sarcoma [12].
The pharmacokinetic (PK) profile of lurbinectedin showed dose linearity at the dose range explored (from 0.02 to 5.0 mg/m2), with no significant relationship between lurbinectedin clearance (CL) and body surface area (BSA) [6].
Based on the proven activity of lurbinectedin in non-Japanese patients, the clinical development of lurbinectedin was initiated in Japan with this phase I study to determine the RD and suitable schedule for the next phase II studies in Japanese patients with unresectable and advanced solid tumors.
Materials and Methods
This clinical trial was conducted in Japan in compliance with ICH Good Clinical Practice guidelines and the “Ministerial Ordinance on the Standards for the Implementation of Clinical Studies on Pharmaceutical Product” (GCP) in Japan. The study protocol was approved by local ethics committee of each study center. Written informed consent was obtained from each patient before the studyspecific procedures.
Inclusion Criteria
Eligible patients were Japanese patients aged ≥ 20 years, with advanced and/or unresectable solid tumors; life expectancy ≥ 3 months; who had recovered from previous toxicities to grade ≤1 (excluding alopecia and non-painful peripheral sensory neuropathy), with ≥1 previous treatment lines but not >3; Eastern Cooperative Oncology Group (ECOG) performance status score ≤ 2; adequate bone marrow, hepatic and renal function; and measurable disease according to the Response Evaluation Criteria In Solid Tumors (RECIST) v.1.1 (Japanese translation version by Japan Clinical Oncology Group [JCOG]), and with documented disease progression.
Exclusion Criteria
Patients were excluded if they had been pretreated with trabectedin; had colorectal cancer or central nervous system primary tumors; had received ≥3 prior chemotherapy lines for advanced/unresectable disease, other recent antitumor therapies (e.g., chemotherapy-containing regimen ≤3 weeks, monoclonal antibody-containing therapy ≤4 weeks or any other anticancer therapy ≤2 weeks, all with respect to study treatment start) or bone marrow and/or stem cell transplantation; were pregnant or lactating women; or had symptomatic brain metastases or leptomeningeal disease,bone marrow involvement, ongoing chronic hepatopathy, active infection, relevant cardiac disease, external drainage, hematological malignancy (or dyscrasia), bowel sub-occlusion or occlusion, human immunodeficiency virus infection,bleeding diathesis or significant coagulopathy, prior or concurrent invasive malignancy (other than the primary study indication) unless in complete remission for at least 3years, or any other disease interfering with the study outcome.
Study Treatment
Treatment consisted of escalating doses of lurbinectedin administered as a 1-hour i.v. infusion on D1 q3wk with and without primary granulocyte colony-stimulating factor (G-CSF) prophylaxis. G-CSF were administered in a standardized way: 3.6 mg given subcutaneously and starting 24-48 hours after lurbinectedin treatment.
Lurbinectedin was supplied as a lyophilized powder concentrate, reconstituted, and diluted with glucose 5% or sodium chloride 0.9% solution. Antiemetic prophylaxis (dexamethasone 8 mg i.v. and ondansetron 8 mg i.v. dose,or their equivalents) was administered before each infusion of lurbinectedin. Treatment was administered until disease progression, unacceptable toxicity, intercurrent illness precluding study continuation, patient refusal and/or non-compliance with study requirements, treatment delay >21 days (except if clear clinical benefit), or requirement of >2 dose reductions.
From Cycle 2 onwards, treatment was to be delayed if the following criteria were unmet: ECOG performance status 0-2; absolute neutrophil count (ANC) 1.5 ×109/L; platelets 100 × 109/L; hemoglobin 9 g/dL; total (or direct) bilirubin 1.5 × upper limit of normal (ULN); grade 1AST/ALT; plasma albumin ≥3.0 g/dL; calculated creatinine clearance ≥30 mL/min; grade 1 muscular toxicity, grade 1 for other non-hematological drug-related events; and no signs of chronic heart failure. Then, parameters which were unmet were re-evaluated at least every 48-72 hours during the first week, and as clinically appropriate thereafter. A new cycle could only be started upon recovery of these parameters.If recovery to meet criteria was not observed within 21 days, the patient was to be withdrawn from the trial, except for the case of documented objective clinical benefit.
Dose Escalation and Dose-limiting Toxicities
Dose escalation followed a standard 3+3 phase I study design, with cohorts of three patients treated at each dose level. If no dose-limiting toxicities (DLT) occurred during the first cycle in these three patients, escalation proceeded to the next higher dose level. If one of these patients had a DLT in the first cycle, the dose level was expanded to treat at least six patients.
Initial dose escalation was performed with lurbinectedin without primary (G-CSF) prophylaxis, and included three dose levels: 1.5 mg/m2, 2.5 mg/m2 and 3.2mg/m2. The RD (primary endpoint of the study) was defined as the highest dose level at which less than onethird of evaluable patients had DLTs in Cycle 1. Once the RD for lurbinectedin without primary G-CSF prophylaxis was defined as 2.5 mg/m2, a second dose escalation was continued with the administration of primary G-CSF prophylaxis.The starting dose of lurbinectedin with primary GCSF prophylaxis was the maximum tolerated dose (MTD) (3.2 mg/m2) obtained during the initial dose escalation (lurbinectedin without G-CSF) and was escalated to 3.5 mg/m2; in both dose escalations, the same rules were applied. Once the RD of lurbinectedin with G-CSF prophylaxis was defined (3.2 mg/m2), dose escalation was ceased.
The following DLTs were defined: grade 4 neutropenia (ANC <0.5 × 109/L) ≥3 days; grade 4 thrombocytopenia (platelet count <25 × 109/L); grade ≥3 neutropenia or thrombocytopenia lasting ≥ one week; grade ≥3 febrile neutropenia (or neutropenic infection or sepsis); grade 4 anemia; grade 4 alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) increase irrespective of duration (or grade 3 if lasting for ≥one week); treatment-related grade ≥ 2 ALT or AST increase concomitant with total bilirubin increase ≥2 x ULN and normal alkaline phosphatase (AP); any other grade ≥3 non-hematological AE that is suspected to be related to study drug (except for nausea/vomiting, unless a patient is receiving an optimal antiemetic regimen), hypersensitivity reactions, extravasations,grade 3 fatigue lasting less than three days, and non--clinically relevant isolated biochemical abnormalities; delay in the administration of a subsequent cycle exceeding five days from the treatment due date (i.e., Day 22); toxicities meeting the criteria described above but with delayed onset (i.e., those occurring after Cycle 1); non-compliance with the intended dose intensity in half or more than half of the evaluable patients at any dose level (in which case switching to an alternative schedule may be considered, if appropriate);and a red blood cell transfusion (if the need for the transfusion was drug-related).
Methods for Data Collection
An electronic data capture system, also known as an electronic case report form (eCRF), was used to collect patient data.
Study Assessments
A complete history and physical examination were conducted, including an ECOG performance status score assessment,both at baseline and at the beginning of each treatment cycle. Hematology and biochemistry tests were performed at baseline, weekly during cycles 1 and 2, and on D1 and D8 (hematology tests) and D1 (biochemistry tests) during subsequent cycles. Total protein, albumin, C-reactive protein (CRP), calcium and coagulation tests were measured at baseline and on D1 of each cycle. Abnormal (grade≥ 3) tests were re-assessed at pre-specified times until recovery to grade ≤ 2. Electrocardiograms were done at baseline and repeated if clinically indicated.
Adverse events (AEs) and laboratory variables were assessed at baseline and during treatment and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) v.4, Japanese translation version by the JCOG (13), and coded using the Medical Dictionary for Regulatory Activities (Med-DRA) v.22.0.
Antitumor activity was evaluated every two cycles according to the RECIST v.1.1 [14], Japanese translation version by JCOG. Overall response rate (ORR) was defined as the percentage of patients with complete response (CR) or partial response (PR). Clinical benefit rate (CBR) was defined as the percentage of patients with CR, PR or stable disease (SD) for ≥ 4 months. Progression-free survival (PFS) was defined as the time from start of the treatment until disease progression or death from any cause, whichever occurred first.
Pharmacokinetic Analyses
Eleven samples were collected from each patient to quantify the lurbinectedin plasma concentrations at baseline and at different times during one week after the first treatment administration. The complete total plasma concentration-time profiles of lurbinectedin were measured using validated liquid extraction methods followed by ultra--performance liquid chromatography-tandem mass spectrometry detection. The lower limit of quantification was 0.1 ng/mL and the calibration range was 0.1 to 50 ng/mL.The within-day and between-day precisions ranged from 2.7 to 12.9% and 5.1 to 10.7%, respectively. The within-day and between-day accuracy (bias) ranged from - 10 to 12% and -5 to 6%, respectively.
Statistical Analyses
SAS® version 9.4 or higher (SAS Institute Inc.,Cary, NC, USA) was used to analyze this dataset. Demographics and baseline characteristics of all patients were summarized and presented by dose level. Continuous variables were presented as summary statistics and categorical variables in the frequency tables. PFS was calculated using the Kaplan-Meier approach. Binomial exact distribution was used to calculate 95% confidence intervals (95%CIs) for categorical variables. Total lurbinectedin plasma concentration-time profiles were analyzed by standard non-compartmental analysis (NCA) using Phoenix® WinNonlin® version 6.3 (Certara L.P. [Pharsight], St. Louis, MO). The individual PK parameters were tabulated and summarized.
Results
Patient Characteristics
Twenty-six patients were treated with lurbinectedin in this study: 15 without primary G-CSF prophylaxis and 11 with primary G-CSF prophylaxis. The baseline patient characteristics are summarized in Table 1.
Lurbinectedin without Primary G-CSF Prophylaxis
Most of the 15 patients (67%) treated at all dose levels were female, with a median age of 52 years (range,38-65 years). The most common primary tumors were biliopancreatic (20% of patients), esophageal, endometrial, and parotid gland cancers (13% each). Adenocarcinoma was the most common histological type (60%). The most common disease sites were the lung and lymph nodes (67% of patients each), liver (33%), and pleura (27%). Median number of lines of prior therapy for advanced disease was 2 (range, 1-3 lines), with cisplatin (53%) as the most common prior agent.
Eight patients were treated at the RD (2.5 mg/m2).Half of these patients were female, with a median age of 52years (range, 38-65 years). Primary tumors were distributed equally across a wide range of organs, and adenocarcinoma was the most common histological type (88%). Median number of lines of prior therapy for advanced disease was 2 (range, 1-2 lines), with cisplatin and doxorubicin (50% each) as the most common prior agents.
Lurbinectedin with Primary G-CSF Prophylaxis
Most of the 11 patients (64%) treated at all dose levels were male, with a median age of 61 years (range, 40-77 years). The most common primary tumors were breast and biliopancreatic cancers (18% each). The most common histological type was adenocarcinoma (55%). The most common sites of disease were the liver (55%), lung (46%), and lymph nodes (36%). Median number of lines of prior therapy for advanced disease was 2 (1-3 lines), and the most common agents of prior therapy were carboplatin, cisplatin,gimeracil, oteracil, paclitaxel, and tegafur (36% each).
Nine patients were treated at the RD (3.2 mg/m2).Of these, 67% were male, with a median age of 61 years (range, 40-77 years). Primary tumors were distributed across a a wide range of organs. Adenocarcinoma was the most common histological type (44%). Median number of lines of prior therapy for advanced disease was 2 (1-3 lines), with carboplatin (44%) as the most common prior agent.
Treatment Administration
Lurbinectedin without Primary G-CSF Prophylaxis
Fifteen patients were treated with three dose levels:1.5 mg/m2, 2.5 mg/m2 and 3.2 mg/m2 (Table 2). Forty--seven treatment cycles were administered at all dose levels(median: 2 cycles per patient; range: 1-10 cycles). At the RD(2.5 mg/m2), 22 treatment cycles were given (median: 2 cycles per patient; range: 1-7 cycles). Median relative dose intensity was 98.3% (range: 65.2-100.6%).
Dose administration was delayed in four of 15 treated patients (27%) (two patients treated at the RD [2.5mg/m2] and two patients the MTD [3.2 mg/m2]) because of grade 2 neutropenia related to the study treatment. Two patients (13%) had dose reduction due to hematological toxicity: one patient had grade 2 neutropenia at the RD, and another patient had grade 4 neutropenia (DLT) at the MTD.
Lurbinectedin with Primary G-CSF Prophylaxis
Eleven patients were treated at two dose levels: 3.2mg/m2 and 3.5 mg/m2 (Table 2). Seventy-six treatment cycles were administered at all dose levels (median 4 cycles per patient; range: 1-20 cycles). At the RD (3.2 mg/m2), 69 treatment cycles were given (median: 4 cycles per patient; range 3-20 cycles). Median relative dose intensity was 96.3% (range: 74.2-99.6%).
Dose administration was delayed in two of 11 patients (18%), both treated at the RD (3.2 mg/m2) due to grade 2/3 anemia related to the study treatment. Three patients (27%) had dose reduction at the RD due to hematological toxicity: grade 4 thrombocytopenia (DLT) in two patients, and grade 2 anemia in one patient. In addition, treatment-related grade 3 drug-induced liver injury (DILI) concomitant with grade 4 ALT/AST increased that occurred in one patient treated at 3.5 mg/m2 (MTD) was considered a late-onset DLT (observed during Cycle 2) and led to dose reduction.
Dose Escalation and Recommended Dose
Lurbinectedin without Primary G-CSF Prophylaxis
All 15 treated patients were evaluable for DLTs.No DLTs occurred at the first two dose levels (1.5 or 2.5mg/m2). However, two of eight patients (25%) treated with 2.5 mg/m2 had cycle delays and dose reduction due to treatment-related grade 2 neutropenia.
Two of the four patients treated at 3.2 mg/m2 had DLTs consisting of grade 4 neutropenia lasting ≥ 3 days in one patient, which led to dose reduction, and grade 3 neutropenia lasting ≥ 7 days without effects on treatment in another patient; as a result, this dose was defined as the MTD (Table 2). The hematological DLTs observed in these two patients were predictable, transient and manageable: one patient continued receiving lurbinectedin treatment and the other discontinued treatment after Cycle 2 due to disease progression.
No DLTs were reported in eight patients treated at 2.5 mg/m2 and, therefore, this dose was declared the RD for lurbinectedin without primary G-CSF prophylaxis given as a 1-hour i.v. infusion q3wk (data presented at the ASCO 2018 Annual Meeting) [15]. Then, dose escalation was further continued with the administration of lurbinectedin with primary G-CSF prophylaxis according to the prescribing guidelines, as stated by the study protocol.
Lurbinectedin with Primary G-CSF Prophylaxis
All 11 treated patients were evaluable for DLTs.The starting dose (3.2 mg/m2) was the MTD reached during the dose escalation of lurbinectedin without G-CSF. Two of the nine patients treated at 3.2 mg/m2 had DLTs consisting of grade 4 thrombocytopenia lasting for 1 to 3 days, which resulted in dose reduction in both cases. These hematological abnormalities were manageable, reversible, and transient (Table 2). At 3.5 mg/m2, one of the two treated patients, a 55-year-old male with duodenal papilla cancer as primary tumor and metastases in the liver, had DLTs: grade 4 ventricular arrhythmia on Day 4, grade 4 neutropenia on Day 8, and grade 4 thrombocytopenia on Day 9 and Day 11 after first lurbinectedin infusion. On Day 3, the patient lost consciousness and was admitted to the intensive care unit (ICU). During monitoring, he had an episode of severe arrhythmia compatible with both Torsade’s de Pointes and ventricular fibrillation. QTc prolongation was not observed.Final diagnosis was grade 4 ventricular arrhythmia. He received intravenous infusion of lidocaine with a defibrillator (200-300 J DC counter) for managing the ventricular arrhythmia, and treatment with noradrenaline, magnesium sulfate, propofol, carbohydrates with potassium and sodium, thiamine disulfide/pyridoxine/cyanocobalamin, and fentanyl. Four days later, his general condition improved and the patient left the ICU. Although some degree of hypo9 JScholar Publishers JJ Oncol Clin Res 2023 | Vol 4: 106 calcemia and myelosuppression, including both neutropenia and thrombocytopenia, were developed, no cardiac safety issues including clinically significant arrhythmia were observed afterwards. The patient did not require any medication or treatment for ventricular tachycardia but remained hospitalized for close ECG monitoring. Grade 4 ventricular arrhythmia lasted for 6 days, improved to grade 2 on Day 10, and was resolved on Day 24, when the patient was discharged from the hospital. Grade 4 ventricular arrhythmia was judged as serious and led to treatment discontinuation.Nevertheless, relationship was considered unknown taking into account that, so far, no cardiac events of this type have been associated with lurbinectedin. Therefore, 3.5mg/m2 was defined as the MTD and 3.2 mg/m2 was declared the RD for lurbinectedin with primary prophylaxis G-CSF given as a 1-hour i.v. infusion q3wk (data presented at the ESMO 2020 Virtual Congress) [16].
Safety
All treated patients were evaluable for safety: 15 patients received lurbinectedin without primary G-CSF prophylaxis and 11 with primary G-CSF prophylaxis. Treatment-related AEs and laboratory abnormalities at all dose levels and the RD are shown in Table 3.
Lurbinectedin without Primary G-CSF Prophylaxis
At the RD (2.5 mg/m2), all treatment-related AEs were grade 1/2, with the most common being nausea (75% of patients), decreased appetite (38%) and constipation (25%). Hematological abnormalities (regardless of relationship) included anemia grade 1/2 (88%), lymphopenia (88%;grade 3 in 63%), neutropenia (63%; grade 3 in 13%),leukopenia (63%; grade 3 in 13%), and grade 1/2 thrombocytopenia (25%). All biochemical abnormalities were grade 1/2, with the most common being increases in ALT (88%) and AST (75%). No treatment-related serious AEs were reported.No treatment discontinuations or deaths occurred due to toxicity.
Lurbinectedin With Primary G-CSF Prophylaxis
At the RD (3.2 mg/m2), all treatment-related AEs were grade 1/2, with the most common being nausea (67%), decreased appetite (67%) and constipation (33%). Hematological abnormalities (regardless of relationship) consisted of lymphopenia (all patients; grade 3 in 44%), anemia (all patients;grade 3 in 22%), thrombocytopenia (78%; grade 4 in 22%), leukopenia (44%; grade 3 in 22%), and neutropenia (33%; grade 3 and 4 in 11% each). The most frequent biochemical abnormalities were increases in AP (all patients; grade 3 in 11%), ALT (78%; grade 3 in 22%) and AST (56%;grade 3 in 11%). Two treatment-related serious AEs were reported in two patients treated at the MTD (3.5 mg/m2):grade 3 DILI (late-onset DLT concomitant with grade 4 ALT/AST increased), and grade 4 ventricular arrhythmia (DLT concomitant with other DLTs of grade 4 neutropenia and grade 4 thrombocytopenia). Both serious AEs were finally resolved. Treatment discontinuation due to grade 4 ventricular arrhythmia related to lurbinectedin occurred only in one patient treated at 3.5 mg/m2. No deaths occurred due to toxicity.
Efficacy
All 26 treated patients were evaluable for efficacy:15 patients with lurbinectedin without primary G-CSF prophylaxis and 11 with lurbinectedin with primary G-CSF prophylaxis (Table 4). A swimmer plot depicting individual PFS of all patients treated with and without primary G-CSF prophylaxis by dose level is provided in Figure 1.
Lurbinectedin without Primary G-CSF Prophylaxis
one patient with metastatic breast cancer treated at the RD (2.5 mg/m2) achieved a PR that lasted for 4 months; ORR was 12.5% (95%CI, 0.3-52.6%). Stable disease (SD) was observed in one patient treated at 1.5 mg/m2, three treated at 2.5 mg/m2 (RD), and two treated at 3.2 mg/m2 (MTD). Of these, one patient with sigmoid cancer treated at the MTD had prolonged disease stabilization (SD ≥4 months).Clinical benefit rate at the RD was 12.5% (95%CI,0.3-52.6%) and median PFS was 1.3 months (95% CI, 0.6-2.6 months).
Lurbinectedin with Primary G-CSF Prophylaxis
no objective responses were reported. All 11 patients had SD as best response; three prolonged disease stabilizations (SD≥4 months) were observed among the nine patients treated at the RD (3.2 mg/m2). Clinical benefit rate at the RD was 33.3% (95%CI, 7.5-70.1%) and median PFS was 6.7 months (95% CI, 2.3-13.7 months).
Pharmacokinetics
All patients were sampled for PK analysis and were suitable for non-compartmental Analysis (NCA). Individual lurbinectedin total plasma concentration-time profiles grouped by dose level are shown in Figure 2. Parameters obtained for lurbinectedin at each dose level are shown in Table 5.
The mean (standard deviation) of total body clearance (CLt) at dose levels of 1.5, 2.5, 3.2 and 3.5 mg/m² was of 8 (1.9), 9.9 (4), 15 (7.4) and 11 (2.3) L/h, respectively. Total body clearance of lurbinectedin in patients treated with 3.2 mg/m² was the highest among the four dose levels tested, most likely due to low levels of alpha-1-acid glycoprotein (AAG) and C-reactive protein (CRP), which are known to correlate with high lurbinectedin CLt, rather than to dose-dependent PK of lurbinectedin. The relationship between CLt and those covariates was confirmed in the present study (Table 5).
Median values of plasma total plasma dose-independent PK parameters obtained in this study were compared with those from the Fist-in-Human (FiH) trial (PM1183-A-001-08 study) in non-Japanese patients at its RD of 7.0 mg (FD) (Table 6). The PK profile of lurbinectedin in Japanese patients appeared to be similar to that observed in non-Japanese patients.
Discussion
This FiH dose-finding study of lurbinectedin defined the RD for phase II studies of lurbinectedin in Japanese patients at 3.2 mg/m2 on D1 q3wk with primary G-CSF prophylaxis.
\The safety profile of lurbinectedin at this RD (3.2mg/m2) with primary G-CSF prophylaxis was predictable.Myelotoxicity was common but reversible and manageable.Non-hematological toxicities were mild/moderate with the most common being nausea and decreased appetite (67% each). Severe myelotoxicity consisted of grade 3 lymphopenia (44%), grade 3 leukopenia, grade 3 anemia, grade ≥3 neutropenia and grade 4 thrombocytopenia (DLT) (22%,each). The most frequent biochemical abnormalities were increases in AP and transaminases, being mainly grade 1/2,with grade 3 ALT (22%) and grade 3 AP/AST (11% each).No deaths or discontinuations due to toxicity occurred at this RD, therefore supporting an acceptable safety profile. As expected, severe myelotoxicity and some non-hematological toxicities at the RD agrees with those observed in prior studies with lurbinectedin when administered alone at 3.2mg/m2 as 1-hour i.v. infusion q3wk in non-Japanese patients with solid tumors [7,8].
Administration of primary G-CSF prophylaxis allowed continuation with lurbinectedin dose escalation to 3.2 mg/m2, which was the MTD reached during dose escalation without primary G-CSF prophylaxis. The use of primary G-CSF prophylaxis reduced the incidence of neutropenia (all grades) to 36% versus 60% with lurbinectedin withoutG-CSF, and allowed patients to receive a higher number of cycles: the median of cycles per patient was 4.0 (range,3-20) with G-CSF versus 2.5 (range, 1-10) without G-CSF. No cases of febrile neutropenia occurred in this trial, even in the patients treated with lurbinectedin without primary G-CSF prophylaxis.
Therefore, the use of primary G-CSF prophylaxis allowed a RD of lurbinectedin 3.2 mg/m2 to be reached, as defined for non-Japanese cancer patients in US and Europe studies where primary G-CSF support was not required [6-8].
Some evidence of antitumor activity was observed with one partial response and three patients treated at the RD (3.2 mg/m2) showing prolonged stable disease in three different tumor types: oropharynx, retroperitoneum and ureter. Of note, median PFS with lurbinectedin with G-CSF (6.7 months) was longer than without G-CSF (1.3 months).However, the number of patients assessed was limited, and further evaluation on efficacy is needed in focused phase II studies.
As result from PK analysis, CLt of lurbinectedin at 3.2 mg/m² was slightly higher than the others dose levels tested. The observed differences in CLt among dose levels (1.5 and 2.5 mg/m² vs. 3.2 and 3.5 mg/m²) could be due to differences in AAG and CRP levels, rather than to dose-dependent PK of lurbinectedin. In fact, such inverse relationship of AAG and CRP plasma levels and direct relationship of albumin with lurbinectedin CLt, has been already detected in pooled phase I/II trials in non-Japanese patients with solid and hematologic malignancies [17]. In addition, from comparison of the lurbinectedin PK parameters obtained from this study with those in FiH study conducted in overseas population [6], pharmacokinetic profile in Japanese patients appeared to be similar to that in non-Japanese patients, although data was limited.
Conclusion
Lurbinectedin administered at 3.2 mg/m2 on D1 q3wk with primary G-CSF prophylaxis showed an acceptable, predictable and manageable safety profile with hints of activity in Japanese patients with unresectable and advanced solid tumors. This dose, 3.2 mg/m2, is the same approved in Western countries, but for Japanese patients, primary G-CSF prophylaxis has to be used. Therefore, this schedule, including G-CSF support, is recommended for future phase II studies conducted in Japanese cancer patients.
Acknowledgments
We gratefully acknowledge the patients, their families and investigator teams for their participation in this phase I trial.
Funding statement
The study was funded by PharmaMar, S.A.
Conflicts of Interest Statement
Shunji Takahashi has received all support for the present manuscript (e.g., funding, provision of study materials,medical writing, article processing charges, etc.) from PharmaMar. He has also received grants or contracts from Daiichi Sankyo, MSD, Bayer, Bristol Myers Squib, Taiho, Eisai,Chugai, Astrazeneka, and Ono pharmaceutical. In addition,he has received personal payment or honoraria for lectures,presentations, speakers bureaus, manuscript writing or educational events from Daiichi Sankyo, MSD, Bayer,Bristol Myers Squib, Taiho, Eisai, Chugai, Astrazeneka, and Ono pharmaceutical. Toshihiko Doi has recived grants or contracts from Lilly, MSD, Daiichi Sankyo, Sumitomo Dainippon, Taiho, Novartis, Merck Biopharma, Janssen Pharma, Boehringer Ingelheim, Pfizer, BMS, Abbvie, Eisai,IQVIA, Chugai Pharma, SHIONOGI and PharmaMar. He has also received personal consultation fees from Sumitomo Dainippon, Taiho, Takeda, Chugai Pharma, Abbvie, Bayer,Rakuten Medical, Otsuka Pharma, KAKEN Pharma, KYOWA KIRIN, SHIONOGI, PRA Health Science, A2 Health Care and Noil Immune. In addition, he has received personal payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from BMS, Rakuten Medical, Ono Pharma, Daiichi Sankyo and AstraZeneca. Furthermore, he has participated on a Data Safety Monitoring Board or Advisory Board from MSD,Daiichi Sankyo, Amgen, Novartis, Boehringer Ingelheim,Janssen Pharma, Abbvie, Bayer and Astellas Pharma.Toshio Shimizu has received grants or contracts as Principal Investigator of clinical trials from PharmaMar, Eli Lilly,LOXO Oncology, AbbVie, Novartis, Daiichi-Sankyo, Bristol-Myers Squibb, Eisai, AstraZeneca, Takeda Oncology, Incyte,Chordia Therapeutics, 3D-Medicine, Symbio Pharmaceuticals,Pfizer and Astellas Pharma. He has also received consulting fees from AbbVie, Daiichi-Sankyo and Chordia Therapeutics; payment or honoraria for lectures, presentations speakers bureaus, manuscript writing or educational events from MSD, Chugai, Eli Lilly and ONO Pharmaceuticals;and support for attending meetings and/or travel from Eisai. In addition, he has received honoraria for participation on Data Safety Monitoring Board and/or Advisory Board from Chordia Therapeutics, AbbVie, Daiichi-Sankyo, Chugai, CMIC and IQVIA. José Antonio López-Vilariño,Carmen Kahatt, Mariano Siguero, Rubin Lubomirov and Ali Zeaiter report personal fees for salary as full time employee and stock ownership from Pharma-Mar, outside the submitted work. Lola Montilla report personal fees for salary as full time employee from PharmaMar, outside the submitted work.
Authorship Contributions
Shunji Takahashi: Conceptualization, Investigation,Resources, Writing - original draft, Writing - review & editing. Toshihiko Doi: Investigation, Resources, Writing -review & editing. José Antonio López-Vilariño: Conceptualization,Methodology, Writing - original draft, Writing -review & editing, Supervision. Carmen Kahatt: Conceptualization,Methodology, Writing - review & editing, Supervision.Mariano Siguero: Methodology, Formal analysis,Writing - review & editing. Rubin Lubomirov: Conceptualization,Methodology, Formal analysis, Writing - originaldraft, Writing - review & editing, Supervision. Lola Montilla:Methodology, Writing - original draft, Writing - review& editing. Ali Zeaiter: Methodology, Writing - review & editing, Supervision. Toshio Shimizu: Investigation, Resources, Writing - review & editing.
Trial Registration Number:
Japan Pharmaceutical Information Center (JAPIC) ID: JapicCTI-184185
Preliminary Results were Presented At-The American Society of Clinical Oncology (ASCO) 2018 Annual Meeting. “Takahashi S, Shimizu T, Doi T, Lopez-Vilariño JA, Nuñez R, Kahatt CM, et al. Phase I trial of lurbinectedin (PM1183) in Japanese patients with advanced tumors: Results of the dose escalation part. Journal of Clinical Oncology. 2018;36(15_suppl):2551.
-The European Society for Medical Oncology (ESMO) 2020 Virtual Congress. “Takahashi S, Shimizu T, DoiT, Lopez-Vilarino JA, Martin RN, Kahatt C, et al. 551P Phase I study of lurbinectedin in Japanese patients with pretreated advanced tumours: Final results. Annals of Oncology. 2020;31:S478”.
- Cuevas C, Perez M, Martin MJ, Chicharro JL, Fernandez-Rivas C, Flores M, et al. (2000) Synthesis of ecteinascidin ET-743 and phthalascidin Pt-650 from cyanosafracin B. Org Lett 2: 2545-8.
- Bueren-Calabuig JA, Giraudon C, Galmarini CM, Egly JM, Gago F (2011) Temperature-induced melting of double-stranded DNA in the absence and presence of covalently bonded antitumour drugs: insight from molecular dynamics simulations.Nucleic Acids Res 39: 8248-57.
- Harlow ML, Maloney N, Roland J, Guillen Navarro MJ, Easton MK, Kitchen-Goosen SM, et al. (2016) Lurbinectedin inactivates the Ewing sarcoma oncoprotein EWS-FLI1 by redistributing It within the nucleus. Cancer Res 76: 6657-68.
- Santamaria Nunez G, Robles CM, Giraudon C,Martinez-Leal JF, Compe E, Coin F, et al. (2016) Lurbinectedin specifically triggers the degradation of phosphorylated RNA polymerase II and the formation of DNA breaks in cancer cells. Mol Cancer Ther 15: 1-14.
- Leal JF, Martinez-Diez M, Garcia-Hernandez V, Moneo V, Domingo A, Bueren-Calabuig JA, et al. (2010) PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity. Br J Pharmacol 161: 1099-110.
- Elez ME, Tabernero J, Geary D, Macarulla T, Kang SP, Kahatt C, et al. (2014) First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors.Clin Cancer Res 20: 2205-14.
- Trigo J, Subbiah V, Besse B, Moreno V, Lopez R, Sala MA, et al. (2020) Lurbinectedin as second-line treatment for patients with small-cell lung cancer: a single-arm, open-label, phase 2 basket trial. Lancet Oncol 21: 645-54.
- Gaillard S, Oaknin A, Ray-Coquard I, Vergote I, Scambia G, Colombo N, et al. (2021) Lurbinectedin versus pegylated liposomal doxorubicin or topotecan in patients with platinum-resistant ovarian cancer: A multicenter, randomized,controlled, open-label phase 3 study (CORAIL). Gynecol Oncol 163: 237-45.
- Singh S, Jaigirdar AA, Mulkey F, Cheng J, Hamed SS, Li Y, et al. (2021) FDA Approval Summary: Lurbinectedin for the Treatment of Metastatic Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 27: 2378-82.
- NCCN (2021) Clinical Practice Guidelines in Oncology.Small Cell Lung Cancer. 11.Dingemans AC, Fruh M,Ardizzoni A, Besse B, Faivre-Finn C, Hendriks LE, et al.(2021) Small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology: official journal of the European Society for Medical Oncology/ ESMO 32: 839-53.
- Subbiah V, Brana I, Longhi A, Boni V, Delord JP,Awada A, et al. (2022) Antitumor Activity of Lurbinectedin, a Selective Inhibitor of Oncogene Transcription, in Patients with Relapsed Ewing Sarcoma: Results of a Basket Phase II Study. Clinical cancer research : an official journal of the American Association for Cancer Research 28: 2762-70.
- Japanese translation of common terminology criteria for adverse events (CTCAE), and instructions and guidelines (2004) Int J Clin Oncol 9: 1-82.
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH,Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228-47.
- Takahashi S, Shimizu T, Doi T, Lopez-Vilarino JA,Martin RN, Kahatt C, et al. (2018) Phase I trial of lurbinectedin (PM1183) in Japanese patients with advanced tumors: Results of the dose escalation part. Journal of Clinical Oncology 36: 2551.
- Takahashi S, Shimizu T, Doi T, Lopez-Vilarino JA,Martin RN, Kahatt C, et al. (2020) 551P Phase I study of lurbinectedin in Japanese patients with pretreated advanced tumours: Final results. Annals of Oncology 31: S478.
- Fernandez-Teruel C, Gonzalez I, Troconiz IF,Lubomirov R, Soto-Matos A, Fudio S (2019) Population-Pharmacokinetic and Covariate Analysis of Lurbinectedin (PM01183),a New RNA Polymerase II Inhibitor, in Pooled Phase I/II Trials in Patients with Cancer. Clin Pharmacokinet 58: 363-74.
FIGURE 1
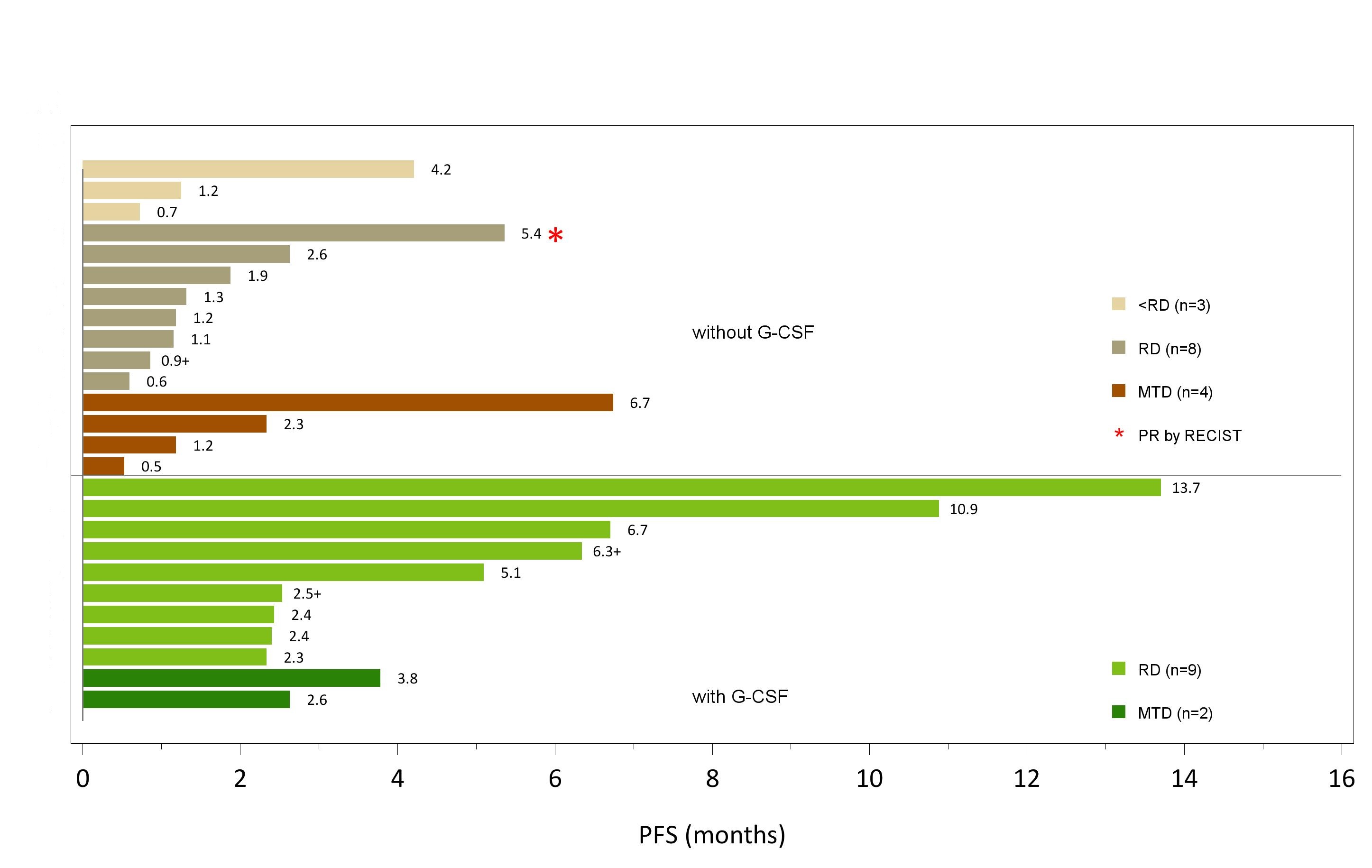
Figure 1: Lurbinectedin swimmer plot of progression-free survival in all individual patients treated with and without primary G-CSF prophylaxis by dose level G-CSF, granulocyte colony-stimulating factor; MTD, maximum tolerated dose; PFS, progression-free survival; PR, partial response; RECIST, Response Evaluation Criteria In Solid Tumors v.1.1 (Japan Clinical Oncology Group translation version); RD, recommended dose.
FIGURE 2
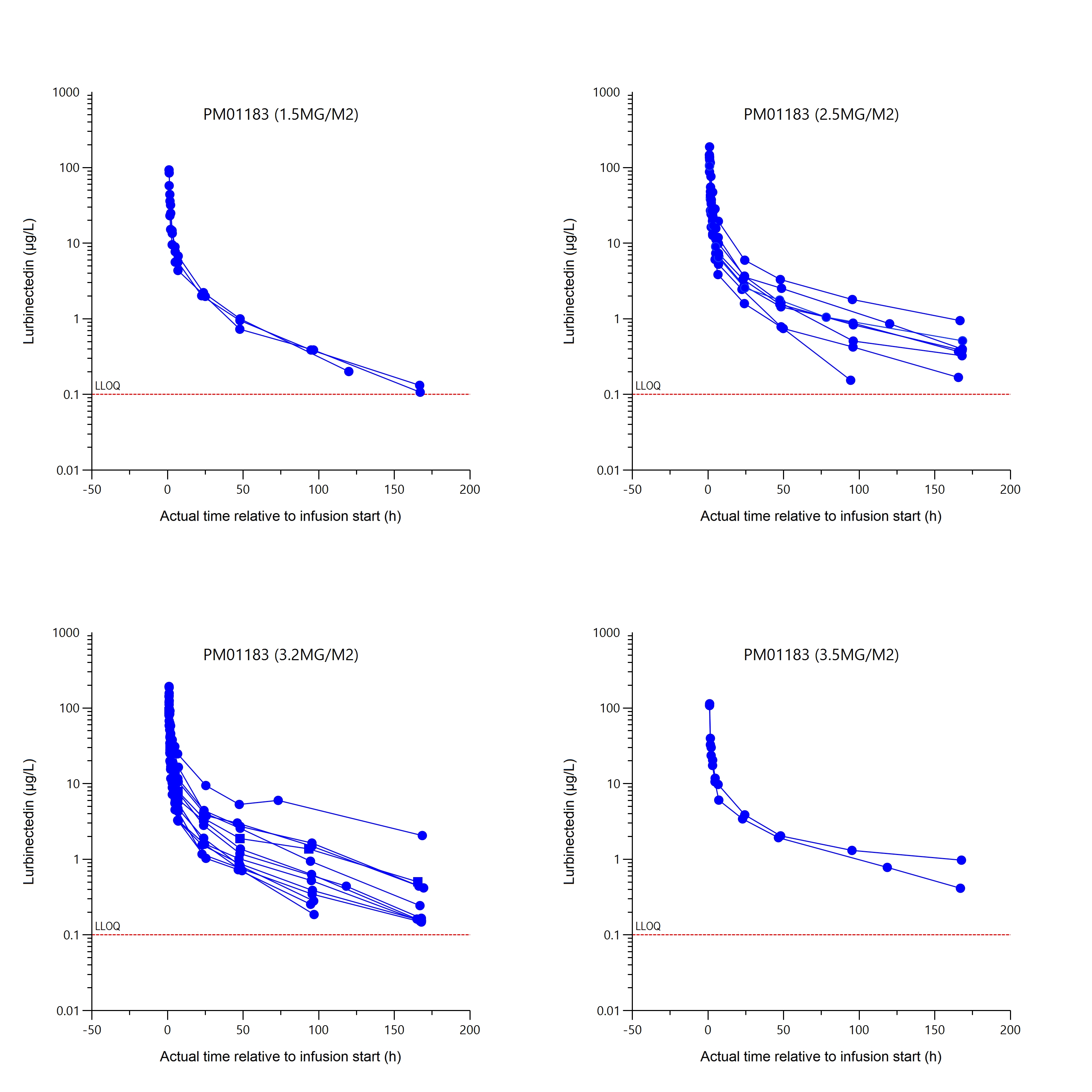
Figure 2: Lurbinectedin total plasma concentration-time profiles by dose level h, hour; LLOQ, lower limit of quantification; PM01183, lurbinectedin.
FIGURE 3
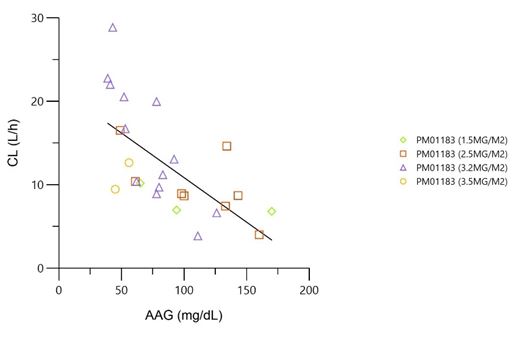
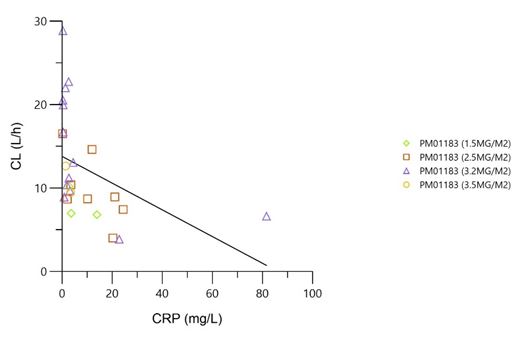
Figure 3: Relationship of lurbinectedin CLt with AAG and CRP by dose level AAG, alpha-1-acid glycoprotein; CLt, total body clearance; CPR, C-reactive protein; PM01183, lurbinectedin; RSQ, coefficient of determination
Tables at a glance
Figures at a glance