Rare Presentation of Megalencephalic Leukoencephalopathy with Subcortical Cysts: A Case Study of Van Der Knaap Disease in a Pediatric Patient
Received Date: February 29, 2024 Accepted Date: March 29, 2024 Published Date: April 01, 2024
doi: 10.17303/jocr.2024.5.101
Citation: Muhammad Tahir Khan, Fariha Shams, Rewati Raman, Ammara Naseem, Muhammad Kaleem (2024) Rare Presentation of Megalencephalic Leukoencephalopathy with Subcortical Cysts: A Case Study of Van Der Knaap Disease in a Pediatric Patient. JJ Oncol Clin Res 5: 1-7
Abstract
Background: Van der Knaap disease is a rare hereditary autosomal recessive ailment that results in macrocephaly, leukodystrophy, and subcortical cysts in 75% of patients due to mutations in the MLC1 gene on the long arm of chromosome 22qtel. A mutation in the MLC1 gene results in a deficiency of ion and water balance in the brain, producing cerebral white matter edema and vacuole development. The MLC1 gene encodes plasma membrane protein present in the brain, spleen, and leukocytes. Clinically, it shows up as extrapyramidal symptoms including stiffness, dystonia, and dysarthria, as well as neurodegenerative symptoms like motor developmental delay, gradual onset of ataxia, and seizures in nearly all patients. The neuroimaging findings, which spare the deep and cerebellar white matter, include megalencephaly, diffuse bilateral inflated appearance of cerebral white matter, and bilateral subcortical cysts with CSF intensity in the temporal and fronto-parietal areas.
Case presentation: A two-year-old female toddler with delayed developmental milestones who also suffers fits. Without any prenatal, natal, or postnatal difficulties, she was born naturally at full term. Her parents claim that at the age of 10 months, she started experiencing generalized tonic-clonic seizures, which lasted between one and two minutes. She was a non-consanguineous marriage's offspring. Upon examination, the anterior fontanelle, other scalp, and ocular examination were all normal. The head circumference was 38 cm, which was above the 97th percentile for her age. Imaging revealed multiple, well-defined, variable-sized intra-axial cysts in place of the subcortical white matter of the bilateral cerebral hemispheres, sparing the basal ganglia, brain stem, and bilateral cerebellar hemispheres, with CSF intensity signals that are hypointense on T1 weighted images and hyperintense on T2 weighted images. Additionally dilated are the ventricular and extraventricular CSF spaces. These imaging characteristics, in addition to the clinical signs, make megalencephalic leukoencephalopathy/Van der knaap illness distinct.
Conclusion: An uncommon inherited autosomal recessive disorder caused by mutations in the MLC1 gene, Van der Knaap disease is highlighted in this case report for its distinctive clinical and neuroimaging features. The MLC1 gene mutation causes cerebral white matter edema and the growth of subcortical cysts by disrupting the equilibrium of ions and water. Clinically, patients display a range of symptoms, including extrapyramidal characteristics and signs of neurodegenerative disorders. The complex nature of this condition is highlighted by the case study of a two-year-old female toddler with generalized tonic-clonic seizures and missed developmental milestones. The neuroimaging findings that distinguish megalencephalic leukoencephalopathy/Van der Knaap disease further include multiple intra-axial cysts in the cerebral hemispheres and spared deep and cerebellar white matter. This case report advances knowledge of the various clinical manifestations and imaging patterns connected to this uncommon illness.
Keywords: Case report; MLC; Megalencephally; Van der Knaap disease; MRI
Introduction
Megalencephalic leukoencephalopathy (MLC), often referred to as Van der Knaap disease with subcortical cysts, is a hereditary condition that causes cerebral white matter edema. As a rare condition, van der Knaap disease is estimated to affect less than 1 in 1,000,000 people. No thorough research has been done to evaluate its prevalence worldwide. Furthermore, there are very few instances of occurrences that happen during the neonatal stage [1]. Megalencephaly, an enlarged brain, and leukoencephalopathy, abnormalities in the white matter, are the main symptoms of this illness [1]. MLC can be divided into two phenotypes: a remitting phenotype and the traditional, deteriorating phenotype. Recessive mutations in the MLC1 gene cause the conventional MLC, also known as MLC1, whereas recessive GLIALCAM mutations cause MLC2A. The dominant GLIALCAM mutations are associated with the remitting MLC or MLC2B [3,7].
In the first year of life, macrocephaly is a common feature of the clinical presentation of the condition, which is often accompanied by neurologic symptoms such as ataxia, spasticity, and epilepsy. Although it is thought that mortality is low, there isn't any particular research on survival. Patients with the improving phenotype display an initial presentation that is comparable to that of typical MLC but with a more favorable clinical trajectory [2].
The MLC1 gene and the HEPACAM gene are primarily linked to the underlying genetic alterations that cause MLC. However, the exact methods by which these genes affect brain function and development are not yet fully understood. Megalencephaly, anomalies in the cerebral white matter, and subcortical cysts, mostly in the anterior temporal region, are all MRI findings in MLC [4].
Van der Knaap disease is extremely rare, so it's difficult to determine its exact global frequency. However, the Agrawal community in India and Turkish communities are more likely to be affected [1]. The illness causes a variety of neurological symptoms, including ataxia, stiffness, seizures, motor developmental delay, and modest cognitive decline. Despite the lack of a specific treatment, clinical signs and MRI results are often used to confirm the diagnosis [6].
Case Presentation
A two-year-old girl with seizures and delayed developmental milestones was brought to the pediatric clinic by her worried parents. She was a product of a non-consanguineous marriage, delivered at full term without any issues throughout the pregnancy, delivery, or postpartum stages. She was born naturally through vaginal birth, and her parents experienced no problems throughout her early years. Her parents claim that she started having seizures at the age of 10 months. The child experiences one- to two-minute-- long generalized tonic-clonic seizure episodes. The parents sought medical help because they were worried about these seizures. On examination, her head circumference was 38 cm, which was greater than the 97th percentile for her age. This finding shows that her head is bigger than normal. The anterior fontanelle, scalp, and ocular examination, however, seem to be unaltered. Antiepileptic drugs were initiated for seizure management, and follow-up visits were recommended to monitor the patient's progress. Despite this, the patient did not attend any follow-up appointments and subsequently did not make any further contact with us, resulting in the loss of patient follow-up data.
MRI Brain
In both cerebral hemispheres, the subcortical white matter was shown to have discrete and varied intra-axial cysts. The basal ganglia, brain stem, and bilateral cerebellar hemispheres were consistently intact despite the cysts' varying diameters (Figure 1). On T1-weighted images, the signal features of these cysts seemed hypointense, however on T2-weighted images, they displayed hyperintense characteristics (Figure 1&2). Additionally, there was dilatation of the ventricular and extraventricular cerebrospinal fluid (CSF) spaces (Figure 1). There were no restriction on diffusion-weighted images (Figure 3).
Discussion
Van der Knaap disease, commonly known as megalencephalic leukoencephalopathy with subcortical cysts (MLC), is a rare neurodegenerative condition with unusual clinical and radiological symptoms [1]. The case discussed here provides insightful information about the symptoms, difficulties with the diagnosis, and consequences of MLC in a child patient
MLC is characterized by disturbances in brain ion and water balance and is brought on by mutations in MLC1 or GLIALCAM [3]. This equilibrium is mediated by the astrocyte-specific membrane protein MLC1, and MLC1 is chaperoned by GLIALCAM to ensure proper location in the astrocytic endfeet [7]. As a result, the MLC1 protein is affected by both the MLC1 and GLIALCAM mutations. Connexin 43 and the chloride channel ClC2 are two other proteins that are transported by GLIALCAM [2]. Contrarily, recessive CLCN2 mutations cause a unique leukoencephalopathy in childhood or adulthood that is characterized by modest motor impairment and retinopathy [4]. Findings from brain MRI reveal myelin micro vacuolization because they show restricted diffusion in particular brain areas [5]. The cerebral white matter signal abnormalities in pediatric patients are diffuse and mild, but the changes in adult-onset cases are more focused. Although enhanced diffusion and significant cerebral white matter abnormalities are seen in MLC, along with myelin macro vacuolization and enlarged extracellular gaps [2]. These findings go against the expectation that GLIALCAM mutations would show symptoms of both MLC1- and CLCN2-associated disorders, even though GLIALCAM is thought to function as a chaperone for both MLC1 and ClC2. In particular, it is discovered that MLC2A closely resembles MLC1 and does not exhibit MRI signs of CLCN2-related illness. We still don't fully understand how MLC1, GlialCAM, and ClC2 interact with one another to regulate ions and water in the brain.
Delayed developmental milestones, seizures, and distinctive MRI abnormalities, such as subcortical cysts in the cerebral white matter, are the defining characteristics of MLC. In our case, the patient, a child, experienced generalized tonic-clonic seizures and delayed developmental milestones, both of which were consistent with the range of MLC presentations that have been described in the literature [1].
The MRI results in this instance closely match the traditional radiological characteristics linked to MLC. The basal ganglia, brain stem, and cerebellar hemispheres were not affected by the many, clearly defined intra-axial cysts because they were present in the subcortical white matter of the cerebral hemispheres [2]. These cysts' T1 and T2-weighted imaging signal characteristics further support the distinctive MRI pattern seen in MLC instances [3]. Additionally, the ventricular and extraventricular CSF spaces are dilated, which is consistent with the radiological alterations that have been observed in MLC [4].
The patient's non-consanguineous origin in this instance highlights the complex genetic foundation of MLC. Classic MLC has a well-established autosomal-recessive inheritance pattern, with mutations in genes like MLC1 and GLIALCAM being implicated [5]. Further research is needed to determine the precise genetic abnormalities that cause MLC symptoms, especially in cases resulting from non-consanguineous unions.
Due to the rarity of MLC, thorough diagnostic procedures and a high index of suspicion are required. Genetic testing is necessary for a conclusive diagnosis, and it may inform genetic counseling for affected families, even if the clinical presentation and distinctive MRI findings frequently arouse suspicions. For a precise diagnosis and effective treatment plans, a multidisciplinary approach comprising pediatric neurologists, radiologists, and geneticists is essential.
With a focus on treating symptoms and offering suitable medications for epilepsy control, MLC management continues to be mostly supportive. To comprehend the pathophysiological mechanisms underlying the disease and to investigate potential targeted therapies, research is still being done [6].
Conclusion
In conclusion, this work sheds light on the unique Van der Knaap disease presentation in a pediatric patient, providing knowledge on the disease's hereditary background, clinical variety, and distinctive MRI features. The importance of MLC1 and GLIALCAM mutations in brain ion and water control is emphasized, and the need for individualized treatment is made clear by the vast range of clinical severity. Despite the observational aspect of the study's design's shortcomings, the large cohort size more than makes up for them by giving a complete picture of the condition. Early diagnosis may be possible thanks to the study's emphasis on MRI characteristics that distinguish between MLC variations. In the end, this research encourages future investigation into the subtleties of Van der Knaap disease by adding critical knowledge to its understanding and therapy.
Limitations of the Case Report
In the example at hand, genetic testing was not done, which might have revealed important details about the underlying genetic abnormalities. Additionally, the small sample size due to the rarity of Van der Knaap disease may reduce the statistical validity of the results. Last but not least, while efforts have been made to clarify the functions of particular genes, the precise mechanisms and relationships causing the disease remain active areas of investigation and conjecture.
Declarations
Ethics Approval and Consent to Participate
Ethical approval was taken from Dr Nighat Haroon Khan, the Head of Department of Radiology, at Punjab Institute of Neurosciences, Lahore, Pakistan. The patient and his parents signed a written informed consent form. This study was in compliance to the latest version of the Helsinki Declaration.
Consent for Publication
Written informed consent was taken from the patient and his parents for the publication of this case and the relevant radiological images.
Availability of Data and Material
All relevant images have been uploaded along with the manuscript.
Competing Interests
The authors have no competing interests.
Funding
No funding was received for this study.
Authors’ Contributions
MTK conducted literature review, drafted initial document, created images, and amended the final draft. FS and RR oversaw the research and helped with revision. AN and MK revised the manuscript and edited images. The final version of the manuscript was approved by all authors.
Acknowledgements
None
- Al Homsi A, Jhancy M, Chowdhury F, Hossain S, Ahamed R (2020) Van der Knaap Disease (Vanishing White Matter) – Unusual Presentation in a Neonate: A Case Report. Neurology India, 68: 669.
- Hamilton EM, Pinar Tekturk, Cialdella F, Diane, Wolf NI, Cengiz Yalcinkaya, et al. (2018) Megalencephalic leukoencephalopathy with subcortical cysts. Neurology, 90: e1395-403.
- Khan IM, Shabbir A, Sadaf Naz, Zulfqar R (2020) Van Der Knaap Disease in a 3-year-old Male Child: A Case Report. Journal of Islamabad Medical & Dental College, 9: 145-8.
- Saanida MP, Varghese L, Thomas R, Sandeep Govindan Prasad (2021) Siblings with megalencephalic leukoencephalopathy with subcortical cysts van der Knaap disease. BJR case reports, 7: 20200150-0.
- Knaap MS van der, Schiffmann R, Scheper G (2007) Conversion of a Normal MRI into an MRI Showing a Cystic Leukoencephalopathy is not a Known Feature of Vanishing White Matter. Neuropediatrics, 38: 264-4.
- van der Knaap MS, Barth PG, Stroink H, van Nieuwenhuizen O, Arts WFM, Hoogenraad F, et al. (1995) Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Annals of Neurology, 37: 324-34.
- Shukla P, Balakrishnan P, Agarwal N, Ghosh M, Kabra M, Sharma R, et al. (2008) Prenatal diagnosis of megalencephalic leukodystrophy. Prenatal Diagnosis, 28: 357-9.
FIGURE 1
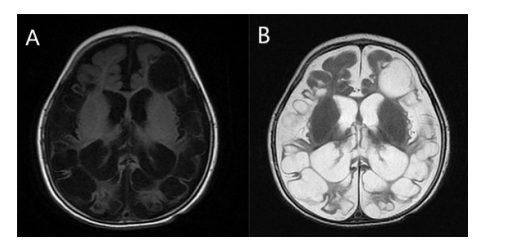
Figure 1: A 2-year-old female child presented with delayed developmental milestones. Axial T1(A) & T2-weighted images (B) showing multiple variable-sized cysts
FIGURE 2
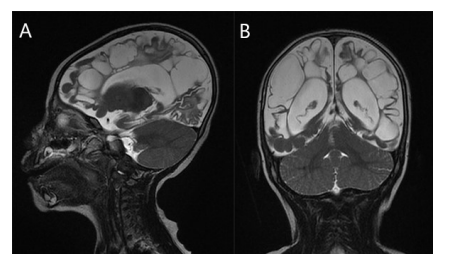
Figure 2: A 2-year-old female child presented with delayed developmental milestones. T2-weighted images sagittal (A) & coronal (B) showing hyperintense multiple intra-axial cysts
FIGURE 3
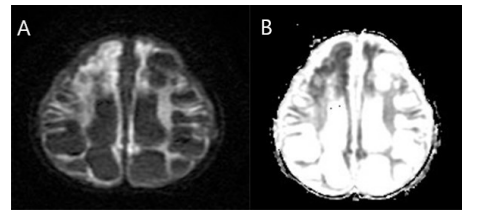
Figure 3: A 2-year-old female child presented with delayed developmental milestones. Diffusion-weighted Weighted Image (A) and apparent Diffusion Coefficient image (B) show no restricted diffusion
Figures at a glance