Synthesis and Biological Evaluation of 2-Substituted-Aryl- 3-Substituted- Phenyl- Oxazole- 4-Thiazolidines as Cyline-Dependent Protein Kinase 2 (CDK2) Inhibitors
Received Date: March 13, 2023 Accepted Date: April 07, 2023 Published Date: April 10, 2023
doi: 10.17303/jocs.2023.1.103
Citation: VM Nikose (2023) Synthesis and Biological Evaluation of 2-Substituted-Aryl- 3-Substituted- Phenyl- Oxazole- 4-Thiazolidines as Cyline-Dependent Protein Kinase 2 (CDK2) Inhibitors. J Org Chem Chem Sci 1: 1-16.
Abstract
A series of diverse heterocycles of 2-amino-4-phenyl thiazole (1a-o), 2-imine substituted phenyl-4-phenyl thiazole (2a-o) and 2- substituted- aryl- 3- substituted- phenyl- oxazole- 4-thiazolidines (3a-o) were prepared with excellent yield. The synthesized compounds were characterized by IR, H1NMR, C13NMR & Mass spectral analysis. Induced-fit molecular docking (IFD) was performed on analogues of 2-Substituted-Aryl- 3-Substituted- Phenyl- Oxazole- 4- Thiazolidines against target protein (PDB ID 3EZR) having natural kinase inhibitor 3-methoxy-4-{3-[4-(4-methylpiperazin-1-yl)-1H-benzimidazol-2-yl]-1H-indazol-6-yl} aniline and synthesized derivatives were carried out against CDK2 using Auto dock tools. Further in vitro studies were evaluated for their antimicrobial and antifungal activities. These heterocycles have been tested against Gram-positive and Gram-negative bacteria and fungi and show outstanding therapeutic activity.
Keywords: 2- substituted - aryl- 3- substituted phenyl- oxazole- 4-thiazolidines; 2-imine; 4 phenyl thiazole; molecular docking; CDK inhibitors; Induced-fit molecular docking
Introduction
In the history of life, heterocyclic compounds have been utilised as colours, pharmaceuticals, and in many commercially important species and their analogues in which one or more ring carbons have been replaced by a heteroatom, such as nitrogen, oxygen, sulphur, phosphorus, silicon, or a metal, among other things. Nitrogen, oxygen, or both are present in the most prevalent heterocyclic systems. The most common and popular methods for preparing heterocyclic compounds is cyclization of suitable compounds. 4-Thiazolidinones are derivatives or Thiazolidine with a carbonyl group at the 4-position substitution is possible at 2, 3 and 5-position [1-2]. The nucleus is also known as the “wonder nucleus” since it produces a variety of derivatives with various biological activity. As a result, thiazolidinone with various substituents has been produced and used as a better medicinal agent in recent years. 4-oxo-thiazolidines are the most widely studied class of chemicals, with antibacterial, anti-inflammatory, anti-HIV, antitubercular, antioxidant, and analgesic properties [3-8]. The nucleus of 4-thiazolidinones has a distinctive role in medicinal actions such as antibacterial, anticancer, antiviral, cardiovascular, antitumor, and CNS depressive.
Experimental
The melting points were measured in uncorrected open capillary tubes. A Perkin-Elmer 157 spectrometer was used to measure IR spectra in KBr pellets. TMS was used as an internal standard in the recording of H1 NMR spectra in a CDCL3 on a Bruker- variah 300MHz FT NMR spectrometer. TLC on silica gel G plates was used to evaluate the purity of the compounds, and exposure to iodine vapours was used to find the spots.
Method of Synthesis
Synthesis of 2-amino, 4-substituted phenyl thiazole
In 100ml of ethanol, substituted acetophenone (0.1mole) and thiourea (0.2mole) were refluxed overnight with Br2 –H2o. Other chemicals were also crystallised from DMF to produce a solid substance.
Synthesis of 2-imine substituted phenyl 4-substituted phenyl thiazole
A mixture of compound I (0.01mole) and substituted benzaldehyde (0.01mole) on discovery in ethanol in presence of Glacial acetic acid. The reaction mixture was refluxed for 12-14hr and resulting solid was washed with ether and crystallized from DMF similarly other compound were also prepared.
Synthesis of 2-[4 oxo-2-substituted-aryl Thiazolidinyl] substituted phenyl thiazole:
In dry benzene, a mixture of compound 2 (0.01mole) and anhydrous ZnCl2 (one pinch) was added drop by drop with stirring, and the mixture was held at room temperature for 3 days before being refluxed for 12 hours. The resultant solid was washed and recrystallized from DMF after the react mixture was filtered and placed on to ice.
Result And Discussion
In light of these findings, various compounds containing 2-amino, 4-substituted phenyl thiazole, and 2-substituted phenyl imine were synthesized. Moiety has been connected to 4-substituted phenyl thiazole, 2-substituted aryl, 3-substituted phenyl thiazole, and 4-thiazolidinones.
Scheme-1 depicts the chemical sequence that leads to the synthesis of required heterocyclic compounds. By reacting substituted acetophenone with thiazole in the presence of Br2 –H2O and ethanol, the starting material 2-amino, 4-substituted phenyl thiazole (1a-o) was created. 2-Amino-substituted phenyl synthesis 2-[4-oxo-2-substituted aryl-Thiazolidinyl] substituted 4-substituted phenyl thiazole (2a-o) The IR, H1 NMR, C13NMR, and Mass spectra of the 2-substituted aryl-3substituted phenyl thiazole 4-thiazoldinones were obtained by reacting 2-amine substituted phenyl, 4-substituted phenyl thiazole with thioglycolic acid and zinc chloride in the presence of benzene.
Biological Studies
Norfloxacine and Griseofulvaline were used as standards in a biological study of thiourea with various acetophenones and (3a-o). The biological activity of compound (1) has been found to be higher than that of the freshly synthesised (3a-o). For antibacterial and antifungal screening, the synthesised compounds were tested against staphylococcus aureus, E. coli, P. vulgaris, A. niger, B. substillis, and Candida albicans at a concentration of 100 mL as shown in Table 1.
Function of CDK Activities
In the protein databank, there exist a lot of CDK2 electronic structures11. Because of the existence of a natural inhibitor in one of the key active sties, 3EZR was chosen. This structure is also comprehensive and error-free. Inhibition of cyclin A and cyclin E-associated cyclin-dependent kinase-2 (CDK2) activity in tumour cells is an effective technique to induce apoptotic cell death via the E2F pathway. The cyclin groove recognition motif (CRM) in the CDK-inhibitory (CDKI) tumour suppressor protein p27 KIP1 was used to develop and synthesise a series of cyclic peptides with biological activity and structural characterization by NMR and X-ray crystallography12 . Inhibitors of cyclin-dependent kinases (Cdks) have been shown to block Cdk7 and Cdk9, which control transcription, in chronic lymphocytic leukaemia cells. Here we studied the novel Cdk inhibitor SNS-032, which exhibits potent and selective inhibitory activity against Cdk2, Cdk7, and Cdk913 Overexpression of cyclooxygenase-2 (COX-2) is frequently observed in several human cancers, including lung, colon, and head and neck. Malignancies are also associated with the dysregulation of cell cycle events and concomitant elevated activity of cyclin-dependent kinases (CDK). CDK2 is a key cell cycle regulatory protein that controls the transition of cells from G1 to S phase. By suppressing CDK2 activity in the H358 human non-small cell lung cancer cell line, we provide many lines of evidence that demonstrate a functional role for CDK2 in IL-1-induced COX-2 expression [14].
FMolecular docking techniques are used to test fifteen molecules of 2-Cyclohexa-1, 5-diphenyl-4-phenyl-[2, 3’] bithiazolyl-4’-one for their ability to inhibit CDK2 (PDB ID 3EZR) enzyme. The goal is to look at the binding energies, different interaction postures, and probable hydrogen bonding of these compounds in order to better understand how efficient they are as CDK inhibitors, specifically CDK2 inhibitors.
Synthesized molecules (Ligand)
Table 4 listed fifteen compounds that were chosen to explore enzyme inhibition with manufactured molecules (also known as ligands).
Receptor enzyme
The electronic structure of CDK2, with PDB ID 3EZR, was chosen as a target protein. The protein file was obtained from an online database. 3-methoxy -4-{3-[4-(4-methylpiperazin-1-yl) -1H-benzimidazol-2-yl] -1H-indazol-6-yl} As a natural inhibitor, aniline17. The chosen enzyme structure was created without any uncertainties, such as missing atoms or amino acids. After removing all heteroatoms (non-receptor atoms like water, ions, and so on), Kollmann charges were assigned. Using AutoDock’s Addsol function, the Solvation parameters were added to the final macromolecule structure18. The natural inhibitory site in an enzyme is considered as the active site of a chosen enzyme and used without further processing.
The binding energies for all docked compounds are listed in Table 4. The docking energies recorded range from –9.68543 to -11.0 653 kcal.mol1, which is higher than natural inhibitor data. The binding energy is not reported for a few compounds. All chemicals have a negative binding energy, which means they can form stable complexes. The development of hydrogen bonding improves the stability of a few molecules (HB). The findings of this study reveal that the current synthetic approach is a straightforward, efficient, and inexpensive way to make biologically active chemical (3a-o), with good results when evaluated at 100 mg Conc. against E. coli, S. aureus, P. vulgaris, A. niger, and C. albicans. Negative binding energies and compact inhibition are reported in molecular docking investigations of fifteen synthesised compounds, 2-Cyclohexa-1, 5-diphenyl-4-phenyl-[2, 3’] bithiazolyl-4’-one compounds. A number of them also mention hydrogen bonding as a possibility. And they’re said to be the best inhibitors since they have greater ligand-enzyme interactions and are more stable. Therefore they show potency to be anti- CDKs agents. Their reported binding energies ranging from -9.68453 kcal. mol−1 to -11.6863 kcal.mol−1 are reported in Table 4. The docking photographs in Table 4 show that the majority of the synthesised molecules bind in active site and remain within the boundaries of the designated active site.
Conclusion
Newly produced chemicals are analysed using elemental analysis, infrared spectra, C13 NRM, and H1 NMR spectra. There have been established efficient methods for synthesis of (3a-o) with high yield. It’s also been reported that a molecule with a larger surface area that fits into the active site of a receptor enzyme has a lower binding energy value than a molecule with a smaller surface area.
Acknowledgement
The authors are thankful to Principal Dr. R.S. Bobhate, Vidya Vikas art, commerce and science college, Samudrapur for providing the laboratory facilities.
- Perreux L, Loupy A (2001) A tentative rationalization of microwave effects in organic synthesis according to the reaction medium and mechanistic considerations. Tetrahedron 57: 9199-223.
- Allen S, Newhouse B, Anderson AS (2004) Discovery and SAR of trisubstituted thiazolidinones as CCR4 antagonists.Bioorganic & Medicinal Chemistry Letters 14: 1619-24.
- Athina A Geronikaki, Alexey A Lagunin, Dimitra I Hadjipavlou-Litina, Phaedra T Eleftheriou, Dmitrii A Filimonov,et al. (2008) J Medicinal Chemistry 51: 1601-9.
- Walsh OM, Meegan MJ, Prendergast RM, Nakib TA(1996) Synthesis of 3-acetoxyazetidin-2-ones and 3-hydroxyazetidin-2-ones with antifugal and antifungal and antibacterial activity. Eur J Med Chem 31: 989–1000.
- Abdel-Rahman RM (2001) Chemoselective heterocyclization and pharmacological activities of new heterocycles--areview.Part V-Synthesis of biocidal 4-thiazolidinones derivatives.Bollettino chimico farmaceutico 140: 401-10.
- Shih MH, Ke FY (2004) Syntheses and evaluation of antioxidant activity of sydnonyl substituted thiazolidinone and thiazoline derivatives. Bioorg Med Chem 12: 4633-43.
- Gadre JN, Nair S, Chitre S (2007) Synthesis of some new 4-thiazolidinones and thiazin-4-ones as biologically potent agent, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem 46B: 653-9
- Vigorita MG, Ottanà R, Monforte F (2001) Synthesis and antiinflammatory, analgesic activity of 3,3’-(1,2-ethanediyl)-bis[2-aryl-4-thiazolidinone] chiral compounds. Part 10. Bioorg Med Chem Lett 11: 2791-94.
- https://cdn.rcsb.org/etl/ligand/img/E/EZR/EZR-large.png
- https://cdn.rcsb.org/etl/poseview/img/ez/3ezr/E/EZR/3ezr_EZR.png
- Rose PW, Beran B, Bi C (2011) The RCSB Protein Data Bank: redesigned web site and web services. Nucleic Acids Res 39: D392-D401.
- Andrews MJI, McInnes C, Kontopidis G, Innes L,Cowan A, et al. (2004) Design, synthesis, biological activity and structural analysis of cyclic peptide inhibitors targeting the substrate recruitment site of cyclin-dependent kinase complexes.Org Biomol Chem 2: 2735-41.
- Chen R, Wierda WG, Chubb S (2009) Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 113: 4637-45.
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr (2000) Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs viatrans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14:1649-60.
Table 4
|
||||
|
Surface Dot 3-D structure |
Binding Energy |
H-bond distance |
Amino acids |
1* |
|
-11.6863 |
2.744652 |
955-DEF |
2 |
|
-10.7636 |
|
958- DEF |
3 |
|
-10.0056 |
2.452949 |
145 -ASP |
4 |
|
-10.0197 |
2.633822 |
83-LEU |
5 |
|
-10.0197 |
2.895364 |
145- ASP |
6 |
|
-10.3206 |
2.907496 |
125 -HIS |
7 |
|
-9.84859 |
2.555301, 2.892527, 2.900609A |
14 -THR |
8* |
|
-11.0653 |
2.980913A |
85-GLN,85-LEU |
9 |
|
|
|
|
10 |
|
-9.62396 |
2.59475, |
83-LEU |
11 |
|
-9.62396 |
2.59475, |
83-LEU |
12 |
|
-10.7636 |
--- |
|
13 |
|
-9.68543 |
---- |
|
14 |
|
-10.5341 |
2.840492 |
|
15 |
|
-10.475 |
1.064365 |
145- ASP |
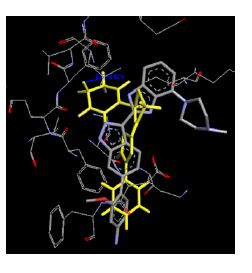
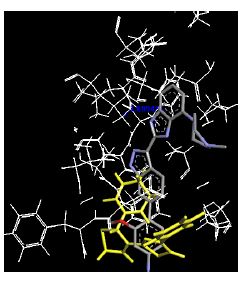
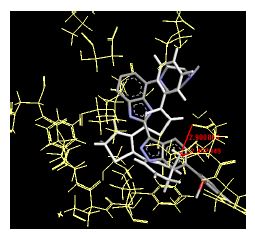
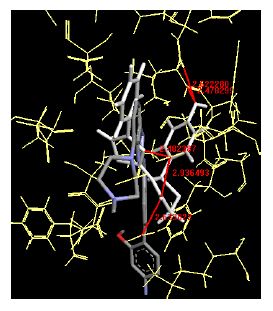
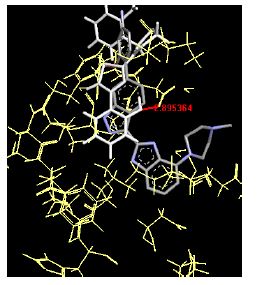
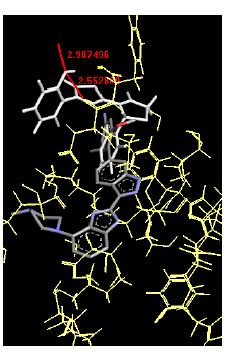
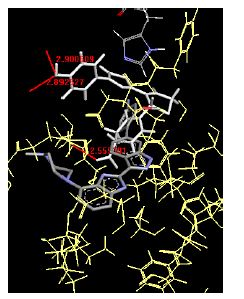
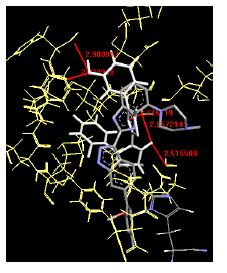
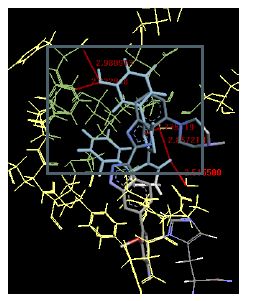
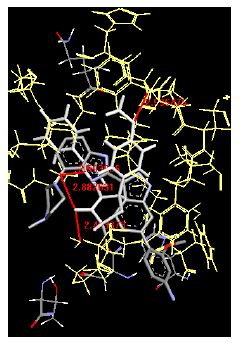
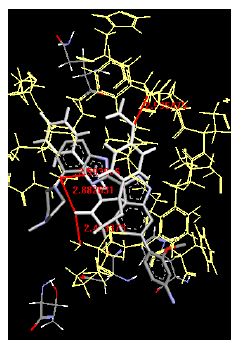
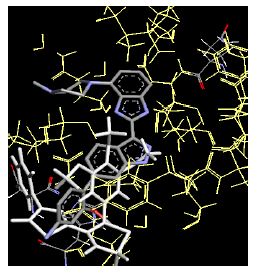
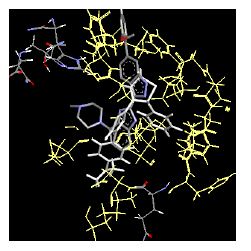
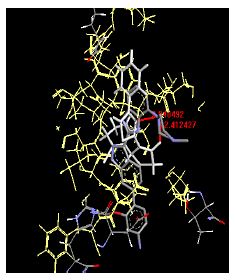
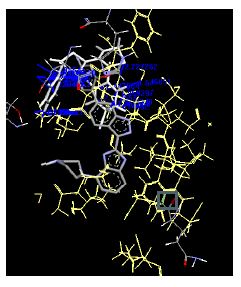
Table 4: Lists obtained binding energies for all docked molecules
FIGURE 1
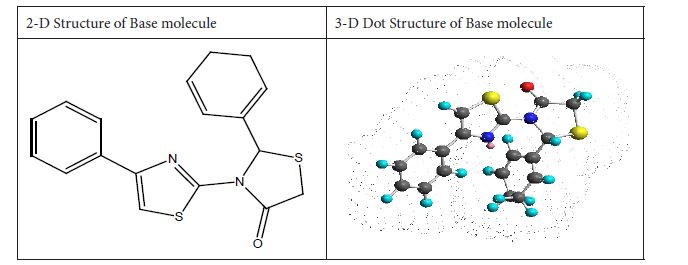
Figure 1: Base molecule 2-Cyclohexa-1, 5-diphenyl-4-phenyl-[2, 3’] bithiazolyl-4’-one
FIGURE 2
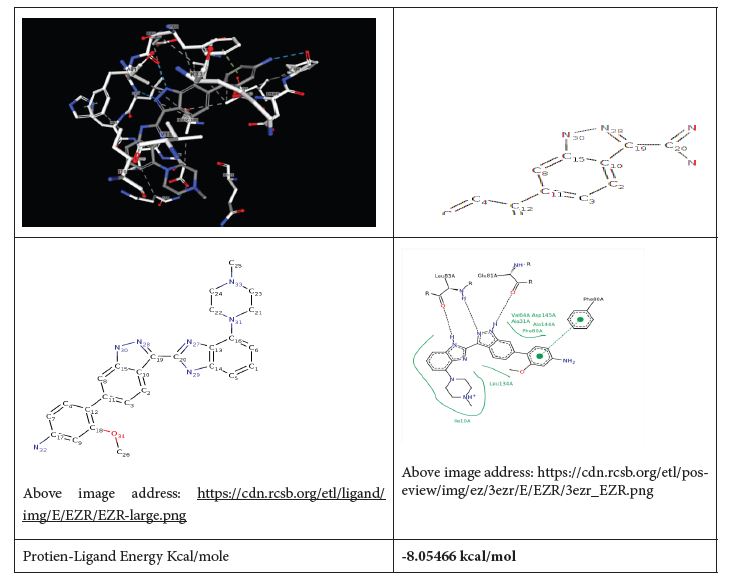
Figure 2: Natural Inhibitor 3-methoxy-4-{3-[4-(4-methylpiperazin-1-yl)-1H- benzimidazol-2-yl]-1H-indazol-6-yl} aniline of PDB ID 3EZR [9,10]
FIGURE 3
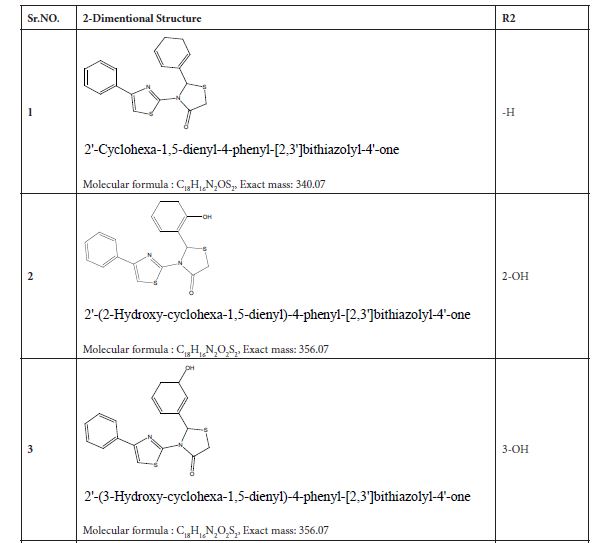
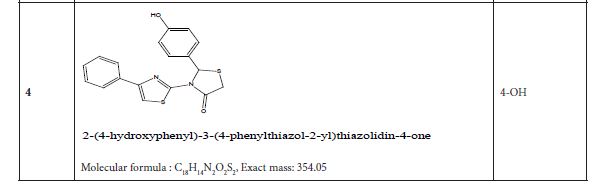
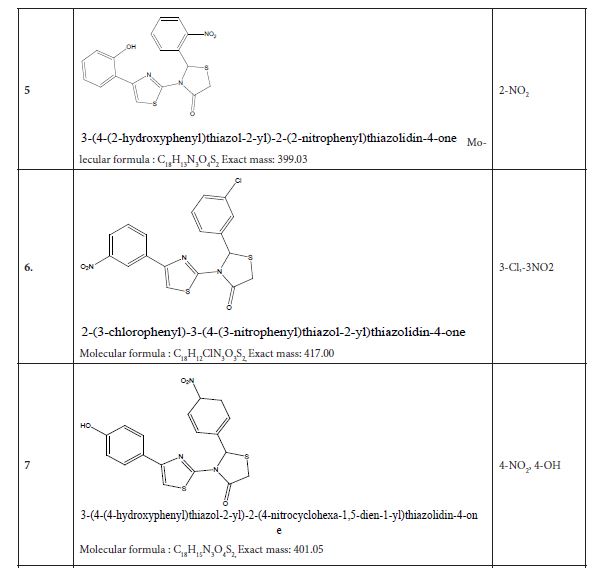
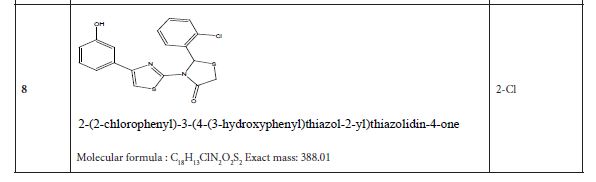
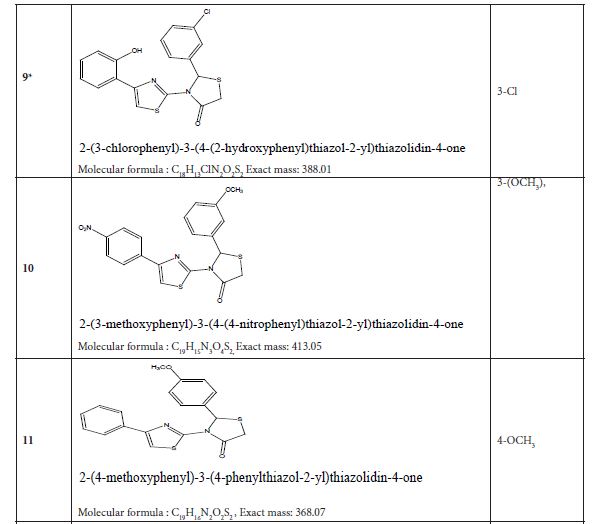
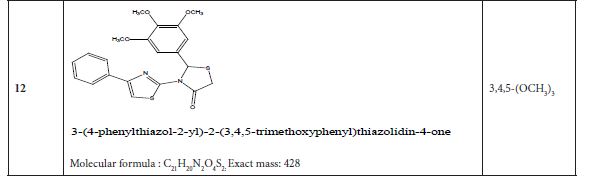
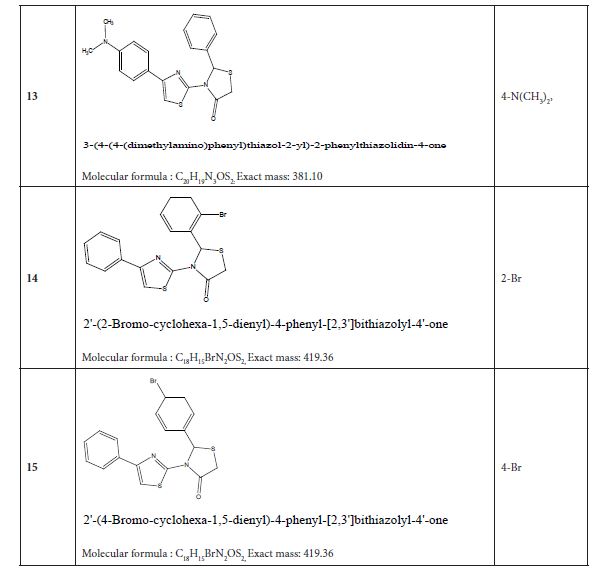
Figure 3: Substituted 2-Cyclohexa-1,5-dienyl-4-phenyl-[2,3] bithiazolyl-4-one
Scheme 1
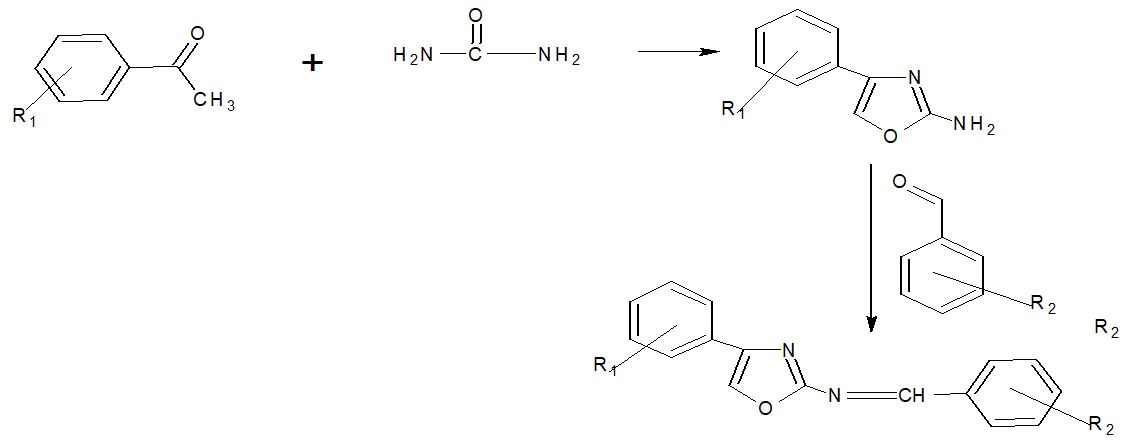
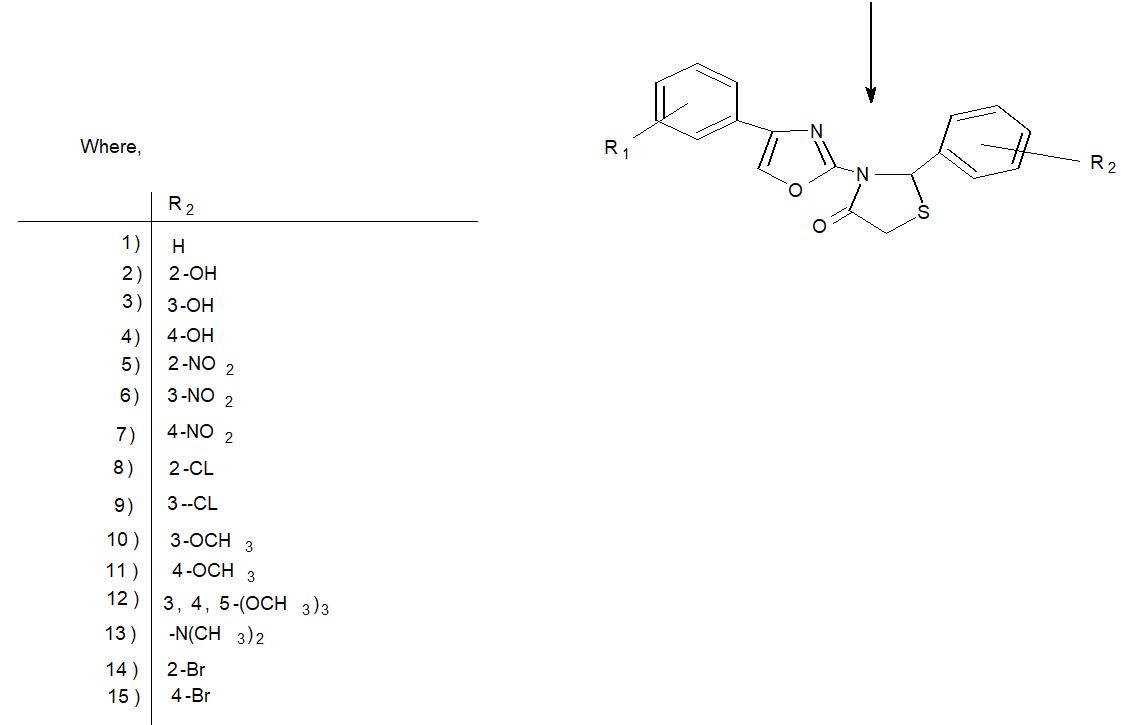
Scheme 1:
Tables at a glance
Figures at a glance