Newly Described Case of Congenital Corneal Opacities in Three Generations of One Family
Received Date: November 17, 2025 Accepted Date: December 04, 2025 Published Date: December 06, 2025
doi:10.17303/jooa.2025.9.202
Citation: Kara R Grimes MD, Jeremy Appelbaum BA, Linda M Reis MS, Elena V Semina PhD, Gerald W Zaidman MD (2025) Newly Described Case of Congenital Corneal Opacities in Three Generations of One Family. J Ophthalmol Open Access 9: 1-4
Abstract
Background: Congenital corneal opacities (CCO) are rare, presenting in ~3 per 100,000 births. Most cases of CCO are isolated without a family history, involvement of siblings, or findings across multiple generations. We describe an unusual autosomal dominant inheritance pattern of CCO affecting six known family members across three generations.
Case Summary: The proband, a 6-month-old boy, presented with bilateral central corneal haze. Four affected family members, including the proband, required penetrating keratoplasty in infancy. Genetic testing via whole exome sequencing of the proband and relatives did not reveal pathogenic variants in genes associated with anterior segment dysgenesis.
Discussion: This case highlights a rare autosomal dominant form of congenital corneal opacity. Reporting this family’s clinical and pathologic findings will increase awareness of this condition, resulting in further testing and possible eventual isolation of the genetic anomaly. This can guide earlier diagnosis, inform genetic counseling, and support timely surgical planning for affected individuals.
Keywords: Congenital Corneal Opacity; Pediatric Corneal Opacity; Inherited Corneal Opacity
Introduction
Congenital corneal opacities (CCO) refer to the clouding or opacification of the cornea present at birth or in early infancy. CCO represent a heterogenous group of disorders with genetic, developmental, or acquired etiologies. Developmental causes are most common and often arise from anterior segment dysgenesis, including Peters anomaly, sclerocornea, Axenfeld-Rieger syndrome, and congenital glaucoma [1]. Acquired causes may result from birth trauma, metabolic disease, or intrauterine infection.
While most cases of CCO occur sporadically, familial forms are occasionally seen. We describe a multigenerational family with an unusual autosomal dominant pattern of congenital corneal opacity, highlighting a phenotype that required early surgical intervention in several affected family members.
Brief Report
A 6-month-old boy was referred for evaluation of bilateral hazy corneas present since birth with progressive worsening. He was born at term without perinatal complications, had an unremarkable neonatal course, and demonstrated normal systemic development with age-appropriate milestones per his pediatrician. Aside from his ocular findings, no systemic abnormalities were noted.
Examination under anesthesia revealed bilateral central corneal haze with 360 degrees of synechiae extending from the iris to the angle and peripheral corneas (Figure 1), resembling an Axenfeld-type adhesion pattern. Corneal haze was present in both eyes, more pronounced in the left. Corneal pachymetry measured 520 m OD and 490 m OS. Ultrasound biomicroscopy and anterior segment OCT confirmed extensive peripheral anterior synechiae. Intraocular pressure (IOP) was 16 mmHg OD and 19 mmHg OS. Anterior chamber depths were 2.06 mm OD and 2.08 mm OS. Red reflexes were present but diminished bilaterally. Both lenses were clear with no evidence of cataract.
Family history revealed that the proband is the third generation and sixth known member of his family to present with bilateral congenital corneal opacities (Figure 2). Two of his older sisters were born with peripheral corneal changes with synechiae but did not require corneal transplant surgery. Their central corneas were clear. The proband’s mother, maternal aunt, and maternal grandfather also had congenital corneal opacities and required corneal transplant surgery during infancy.
At 12-months, the proband underwent corneal transplant surgery in the left eye, followed by the right eye two months later. Pathology of the excised corneal tissue revealed focally absent areas of Descemet membrane, fibrous bands, and attenuated endothelium. After four years of follow-up, the proband has clear corneal transplants in both eyes with good vision. He has no evidence of glaucoma, 4+ red reflex in both eyes, and IOPs of 12 mmHg OD and 11 mmHg OS.
Genetic testing by whole exome sequencing on six affected family members, including the proband, his sisters, mother, maternal grandfather, and maternal half-aunt, and two unaffected maternal aunts did not identify any causative variants.
Discussion
Congenital corneal opacities (CCO) are rare, presenting in approximately 3 of 100,000 births [2]. Most cases of CCO are isolated and sporadic without familial involvement. Therefore, this case affecting six known members across three generations is very rare.
The most common etiology for CCO is Peters anomaly, characterized by central corneal opacity with corresponding defects in the posterior stoma, Descemet membrane, and endothelium. Although this patient presented with central corneal opacities, the autosomal dominant inheritance pattern is uncommon for Peters anomaly. Known autosomal dominant cases of Peters anomaly have been associated with variants in genes such as FOXE3, FOXC1, and PAX6 [3]. These were not present in this family.
The proband was one of four members in this family to require penetrating keratoplasty (PK). In his sisters, the corneal opacity was not severe enough to require surgery. Many patients with CCO typically undergo PK within the first year of life to reduce amblyopia risk, though opacities can clear spontaneously, as in the proband’s sisters. Individuals with CCO will continue to be monitored throughout childhood.
Initial genetic testing via exome sequencing ruled out pathogenic variants in known anterior segment dysgenesis genes. Whole genome sequencing is currently in progress to identify a potential novel gene/mechanism that could provide a clearer understanding of their autosomal dominant condition and inform future genetic counseling. The identification of a specific genetic etiology may facilitate early and accurate diagnosis, enabling timely intervention and potentially improve long-term patient outcomes. The rare clinical and pathological findings observed in this family underscore the importance of reporting such cases, as they contribute to the broader medical understanding of CCO. By sharing this family’s unique case, further research may eventually lead to the isolation and characterization of the underlying genetic anomaly, benefiting not only this family, but also those who may present similarly in the future.
Financial Support
None
Conflict of Interest
No conflicting relationship exists for any author.
Funding
Funding was provided by NIH grant R01EY015518 (EVS).
- Karadag R, Rapuano CJ, Hammersmith KM, Nagra PK (2020) Causes of congenital corneal opacities and their management in a tertiary care center. Arq Bras Oftalmol. 83: 98-102.
- Bermejo E, Martínez-Frías ML (1998) Congenital eye malformations: clinical-epidemiological analysis of 1,124,654 consecutive births in Spain. Am J Med Genet. 75: 497-504.
- Nischal KK (2015) Genetics of Congenital Corneal Opacification--Impact on Diagnosis and Treatment. Cornea. 10:S24-34.
FIGURE 1
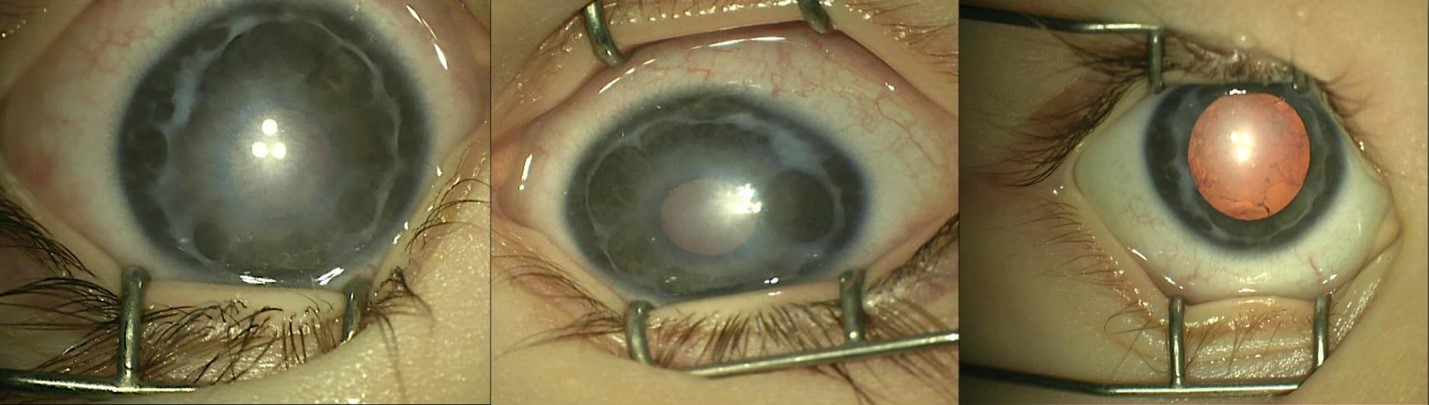
Figure 1: Proband at 6-months of age with Bilateral Central Corneal Haze with 360° of Synechiae from the Angle and the Iris to the Peripheral Cornea.
FIGURE 2
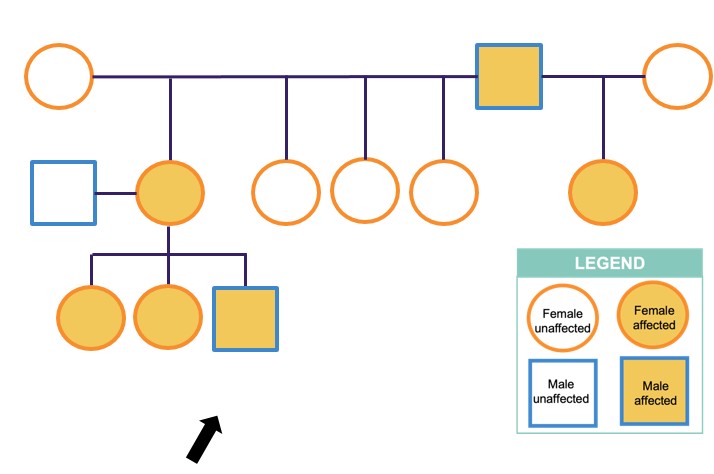
Figure 2: Pedigree of the Proband and Family Members showing Autosomal Dominant Inheritance of Congenital Corneal Opacities
Figures at a glance