Extra Renal Malignant Rhabdoid Tumors in Children: Case Series
Received Date: March 17, 2023 Accepted Date: April 17, 2023 Published Date: April 20, 2023
doi: 10.17303/jpam.2023.3.103
Citation: Yasmine Laraqui, Maryam Cheddadi, Lamia Rouas, Najat Lamalmi (2023) Extra Renal Malignant Rhabdoid Tumors in Children: Case Series. J Pathol Allied Med 3: 1-11
Abstract
Malignant rhabdoid tumor is a rare but high malignant neoplasm associated with a high mortality rate. It is a distinct clinicopathologic entity with a characteristic clinical behavior and pathological features. They usually occur in the kidney but many extra renal rhabdoid tumors were reported; the unifying feature of infantile rhabdoid tumors at all sites is the presence of a mutation or deletion of the INI1 gene located at chromosome 22q11.
We present four cases that demonstrate the importance of early diagnosis to improve the prognostic. The study concerned four children between 6 months and 18 months old, in whom the diagnosis of rhabdoid tumor has been made, localized in retro-auricular, gluteal, pelvic or pleural area.
Keywords: Rhabdoid; Children; Extrarenal
Case reports
First case
We report the case of an 18-month-old male infant,with three weeks ‘history of dry cough, polypnea, and fever without improvement under antibiotic therapy. The chest radiography
showed a left pleural effusion with lysis of sixth rib (Figure 1).
Materials and Methods
Experimental animals
CT showed a large lobulated parietal mass delimiting liquid hypodense areas with pleural effusion measuring 90 × 67× 86 mm. The mass compressed the homolateral lung and mediastinal structures to the right. It was associated with a lysis of the fifth and
sixth ribs with soft tissue extension.
After biopsy, histological examination showed a richly vascularized fibrous tissue composed of large cells ranges with reduced cytoplasm and oval nuclei with vesicular chromatin, and cells ranges with nuclei often eccentric with abundant eosinophilic cytoplasmic inclusion (Figure 2).
Immunohistochemical study revealed a positive staining for cytokeratin, vimentin, epithelial membrane antigen,smooth muscle actin, and CD99. Tumor cells were negative for CD45-CD3- CD20-ALK-CD30, MPO-CD34-CD68-CD1-desmin– myogenin. Also, INI-1 protein labeling was negative (Figure 3).
In front of the clinical deterioration and histopathological results, particularly the loss of expression of INI1,the diagnosis of the RT was retained.
The patient underwent chemotherapy treatment associating vincristine, actinomycin, cyclophosphamide, then cyclophosphamide/Adria and VP16/carboplatin alternately.
Despite all these treatments, there was a deterioration on the patient’s conditions with dependence on oxygen therapy.
The patient died after 2 months and 10 days of diagnosis.
Second case
We report the case of an 18-month-old female infant which presented with a left retroauricular mass, evolving since 5 months and gradually increasing in volume.
Clinical examination highlighted a 2 cm long mass, painless, firm, fixed to the deep plane.
Examination of lymph node areas found some centimetric cervical lymphadenopathy.
Brain CT revealed a malignant process in the left retroauricular soft tissue without endocranial extension.
The histological examination, after excisional biopsy, revealed a malignant proliferation with heterogeneous architecture with lobulated, dense or sometimes alveolar areas.Tumor cells were atypical, polygonal, non-cohesive with often vesicular nuclei, nucleolus, within an eosinophilic cytoplasm of variable abundance. Mitosis is very common.
Faced with this morphological aspect, different diagnoses have been proposed, like a sarcomatous process of the soft tissues, including alveolar rhabdomyosarcoma; a chondrosarcoma because of the lobulated architecture and the vesicular nature of the nuclei;
A PNET (primary neuro-ectodermal tumor).
At first, immunohistochemical study showed a positive labelling for Cytokeratin, EMA, AML and, focally, CD 99 and Wt1. HMB 45 myogenin labeling was negative.
A complementary study was made on the pathology laboratory at Saint Jude Children's Research Hospital in Memphis with INI1 showing loss of its nuclear expression in tumor cells.
The diagnosis of primary malignant rhabdoid tumor (MRT) of the retroauricular soft tissue was thus retained.
The patient underwent a surgical resection of the mass first, for which histological examination confirmed the diagnosis with resection limits less than 1mm. It was followed by chemotherapy and then radiotherapy. The evolution was marked by a local recurrence 10 months later, leading to a second surgery. After follow-up every 3 months, the evolution was favorable.
Third case
A 14 month-old-female was referred to our hospital for a history of a mass in the gluteal region, evolving for 3 months, associated with dysuria. On examination, she was a healthy girl with stable vital signs. Back examination revealed a firm mass with a normal overlying skin.
Ultrasound of the mass revealed uretero-hydronephrosis associated with a left gluteal tissular mass. Surrounding soft tissues were unremarkable.
Hematoxylin-eosin stained sections showed malignant mesenchymal proliferation made up of round or spindle-shaped cells. These cells are small to medium in size, with round or oval hyperchromatic nuclei, with a prominent nucleolus. The cytoplasm is of variable abundance,with imprecise limits, sometimes microvacuolar. The stroma is myxoid.
On immunohistochemistry, neoplastic cells were positive for CD99, EMA, CD56, Vimentine, desmine. There were negative for Cytokeratin, LCA, Bcl6, Myogenin, Myo-D1, PS100 and Synaptophysin. Thus the diagnosis of rhabdoid tumor was made based on morphological and immunohistochemical studies.
The patient was operated on and then lost to follow-up.
Fourth case
6-month-old girl, initially followed in oncology for pelvic neuroblastoma. In the absence of response to chemotherapy,a review was requested.
Histological examination revealed a proliferation of round cells with an abundant eosinophilic cytoplasm and a rounded or oval exentered, hyperchromatic nucleus with a prominent nucleolus. These cells are arranged in plaques on a sometimes myxoid background.
A complementary immunohistochemical study was made objectifying a positive staining for Vimentin,EMA, CD 99, Cytokeratin and CD56 antibodies. Tumor cells were negative for desmin, myogenin and INI 1.
The diagnosis of rhabdoid tumor was thus retained.
The patient underwent a surgical resection of the mass first, followed by chemotherapy and then radiotherapy.The evolution is favorable for now.
Discussion
Malignant rhabdoid tumor is one of the most aggressiveneoplasms in infancy, associated with a reported mortality rate of 80% to 100%. It seems to be confined almost exclusively to children, particularly those less than five years of age [1].
The usual site of occurrence is the kidney but, recently, many other rhabdoid tumors were reported at a number of extra renal sites, particularly extra nervous system,as well as other locations such as the liver, muscle,heart, lung, soft tissues and skin [2].
It was initially associated with other skeletal muscle tumors, but because of the lack of ultrastructural or immunohistochemical proof of myogenic origin, it is now defined as a distinct clinicopathologic entity with a characteristic clinical behavior and histopathologic, immunohistochemical and cytogenetic features [3].
In fact, the establishment of a common genetic deletion on the long arm of chromosome 22 in both renal and extra renal rhabdoid tumors in infants has allowed a unifying approach to the recognition of primary rhabdoid tumors of childhood [2].
At different sites, rhabdoid tumor show similar demographic,clinical and histological features.
These different sites of occurring explain why rhabdoid tumors create a diagnostic challenge.
Clinical presentation is variable, depending on the localization of the tumor. Plus, there are no pathognomonic imaging signs. Nonetheless, imaging may show the heterogeneous nature of the lesion with intense contrast enhancement with necrosis areas and intra tumor calcifications [4].
Thus, the final diagnosis is essentially based on histological,immunohistochemical and cytogenetic features.
Histologically, it is characterized by a diffuse proliferation of “Rhabdoid cells” consisting of monomorphous population of large, relatively noncohesive cells with vesicular nuclei, large nucleoli and glassy eosinophilic cytoplasm with hyaline-like inclusion bodies [5].
However, extra renal rhabdoid tumors are not the only tumors showing these rhabdoid cells, and might be confused with other high grade sarcomas harboring rhabdoid features.
In fact, they are even seen in other soft tissue sarcomas including epithelioid sarcoma, melanoma and rhabdomyosarcomas exhibiting rhabdoid type inclusions [6].
Immunohistochemical studies permit to eliminate differential diagnostic and suggest the diagnosis of rhabdoid tumor. Immunochemistry of rhabdoid tumor shows common mesenchymal and epithelial differentiation.
Vimentin is the most frequently expressed marker,followed by EMA and Cytokeratin.
Epithelioid sarcomas are positive for epithelial markers and vimentine, however they express CD34 which is usually absent in rhabdoid tumors.
Cells with rhabdoid histology may be seen in melanomas but they express PS100, HMB 45 and Melan A, which is lacking in malignant rhabdoid tumors.
Immunohistochemistry permit to eliminate the myogenic origin by the lack immunoreactivity for muscle differentiation with myo D1 and Myogenin [3].
INI 1 immunohistochemical study is now added to the panel in conjunction with the other markers to confirm the histological diagnosis, showing a complete loss of INI 1 protein expression [7].
With cytogenetic and molecular analyzes, the defining feature of ERRTs is an aberration located at chromosome 22q11.1 which encodes the INI 1 gene.
Thus, the histological diagnosis of malignant rhabdoid tumors is based on INI 1 negative expression and the presence of rhabdoid histological features.
Mutations causing inactivation or loss of the INI1 on chromosome 22, aberrations involving 22q11.2 and consequent loss of INI 1 expression by immunohistochemistry are necessary for the diagnosis [8].
The accurate diagnosis of ERRT is important for both correct treatment and prognosis. At the moment, radical surgery remains the only treatment, but may not be realized at advanced stage. Some authors have been reported the beneficial role of chemotherapy and radiotherapy but there is no consistently effective regimen established. Combination of ectoposide and cisplatin or ifosfamide is employed, but the results have not been encouraging [9].
Despite all these treatments, the prognosis of these tumors remains fair to poor. It depends on the stage of the tumor at presentation, the patient’s age at diagnosis, and probably the genetic background. Usually, patients with malignant rhabdoid tumor of soft tissue present at a young age,often with disseminated disease and the aggressive course of the disease is most often fatal with a survival rate varying from 11 to 17 months [10].
Conclusion
Malignant rhabdoid tumor is a rare but high malignant tumor in infant showing diagnostic and treatment challenges. The diagnosis is essentially based on histological, immune histochemical and cytogenetic features. The accurate and early diagnosis is crucial to try to offer the correct treatment and improve the prognostic that remains poor, especially with metastasis present at the time of diagnose.
- Madan K, Bal A, Agarwal R, Das A (2014) Malignant Extra Renal RhabdoidTumour Presenting as Central Airway Obstruction. Case Rep Pulmonol. 27 août e950869.
- Brennan B, De Salvo GL, Orbach D, De Paoli A, Kelsey A et al. (2016) Outcome of extracranial malignant rhabdoidtumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study-EpSSG NRSTS 2005. Eur J Cancer OxfEngl 60: 69-82.
- Oda Y, Tsuneyoshi M (2006) Extrarenalrhabdoid tumors of soft tissue: clinicopathological and molecular genetic review and distinction from other soft-tissue sarcomas with rhabdoid features. Pathol Int 56: 287-95.
- Ivana D, Sofija C, Polina P, Smilja R (2021) Malignant rhabdoid tumor—The great mimicker: Two case reports. Radiol Case Rep. 1 avr 16: 8125-8.
- Gupta RK, Batra VV, Das MC, Sharma A, Narang P (2015) Malignant extra-renal rhabdoid tumor with unusual presentation: A report of two cases. J Cancer Res Ther. déc 11: 963-6.
- Plasschaert H, Deman F, Bempt IV, Labarque V, Aertsen M et al. (2020) ExtrarenalRhabdoidTumour of Soft Tissue. Int J Pathol Clin Res 6: 105.
- Zimmermann A (2016) Malignant Rhabdoid Tumors and Tumors with Rhabdoid Features. In: Zimmermann A, éditeur. Tumors and Tumor-Like Lesions of the Hepatobiliary Tract [Internet]. Cham: Springer International Publishing 1-25.
- Kohashi K, Tanaka Y, Kishimoto H, Yamamoto H, Yamada Y et al. (2016) Reclassification of rhabdoid tumor and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression: three subtypes of rhabdoid tumor according to their histological features.Mod Pathol 29: 1232-42.
- Seeringer A, Bartelheim K, Kerl K, Hasselblatt M,Leuschner I et al. (2014) Feasibility of intensive multimodal therapy in infants affected by rhabdoid tumors - experience of the EU-RHAB registry. Klin Padiatr 226: 143-8.
- Farber BA, Shukla N, Lim IIP, Murphy JM, La Quaglia MP (2017) Prognostic factors and survival in non-- central nervous system rhabdoid tumors. J Pediatr Surg. Mars 52: 373-6.
FIGURE 1
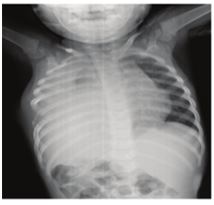
Figure 1: opacity of the left hemithorax compressing heart and the mediastinum structures to the right
FIGURE 2
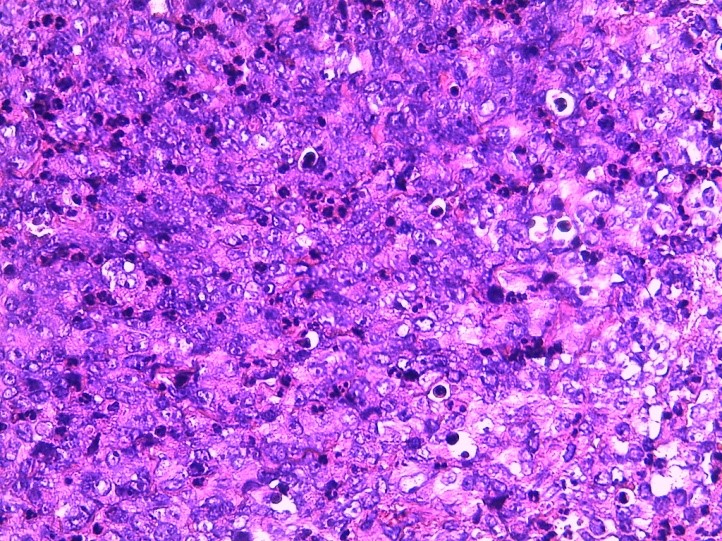
Figure 2: Eosine staining showing large cell ranges with often poorly reduced cytoplasm and oval nuclei with vesicular chromatin, and cell ranges with nuclei often eccentrically placed with abundant eosinophilic cytoplasmic inclusion
FIGURE 3
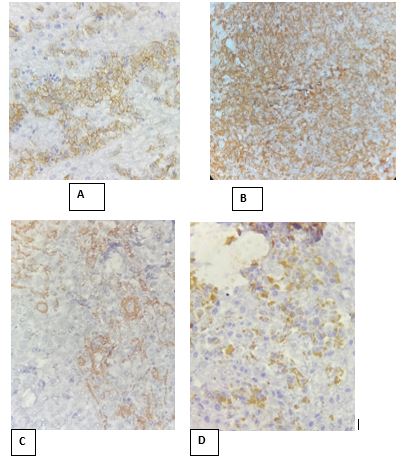
Figure 3: A: Vimentin, B: CD56, C: SMA, D: CKAE1/AE3
FIGURE 4
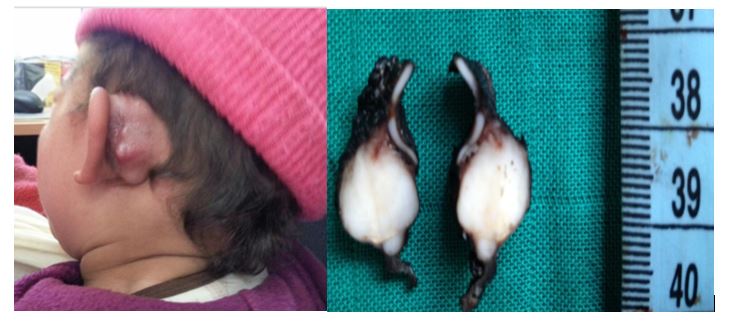
Figure 4: left retro auricular mass
FIGURE 5
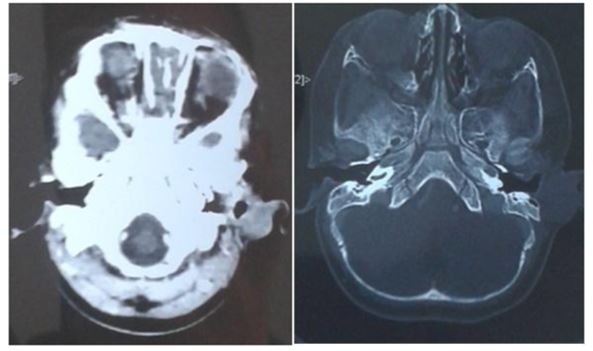
Figure 5: Retro auricular soft tissue process without endocranial extension
FIGURE 6
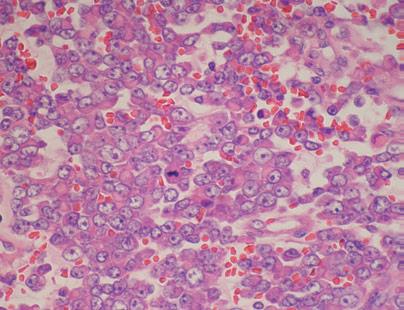
Figure 6: Hematoxylin eosin staining showing atypical rhabdoide cells with vesicular nucleolus nuclei and eosinophilic intra cytoplasmic inclusion
FIGURE 7
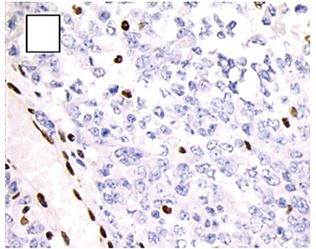
Figure 7: Negative staining with INI1
FIGURE 8
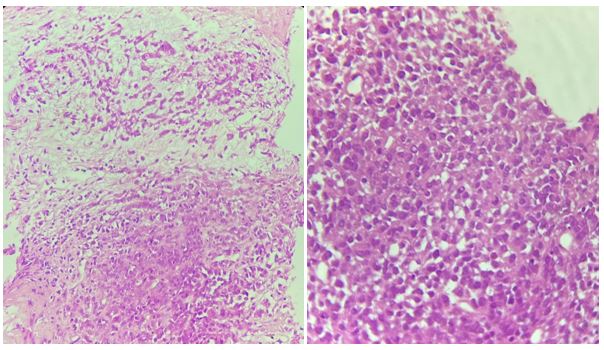
Figure 8: Proliferation of round cells with an abundant eosinophilic cytoplasm and a rounded or oval hyperchromatic and eccentrically placed nucleus arranged in plaques on a myxoid background
FIGURE 9
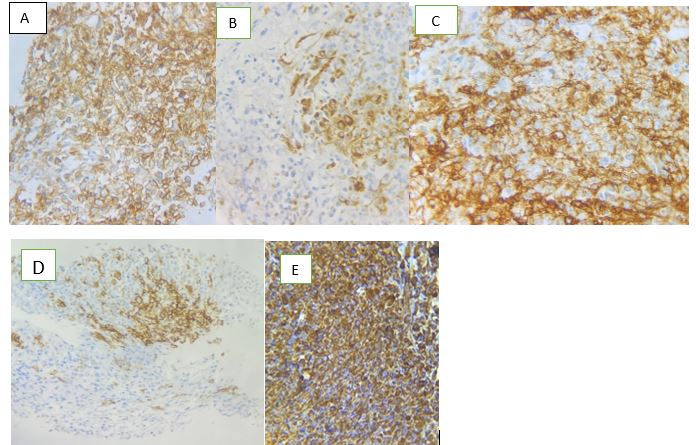
Figure 9: A= CD99+; B=CKAE1/AE3+; C=EMA+; D=CD56+; E= Vimentin+
Figures at a glance