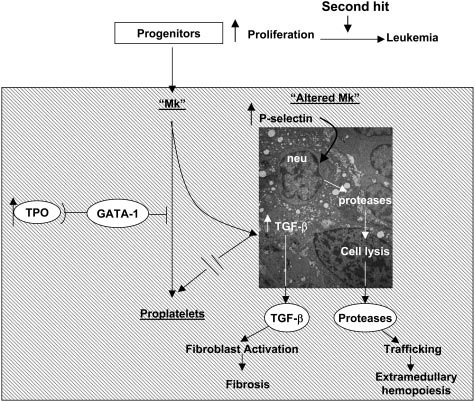
Figure 1. Pathobiologic pathway according to Vannucchi et al linking TPO, GATA-1, and TGF-beta-1 in the development of megakaryocytic myeloproliferation and secondary myelofibrosis in TPOhigh mice followed by endstage myelofibrosis [6].
Figure 1. Pathobiologic pathway according to Vannucchi et al linking TPO, GATA-1, and TGF-beta-1 in the development of megakaryocytic myeloproliferation and secondary myelofibrosis in TPOhigh mice followed by endstage myelofibrosis [6].
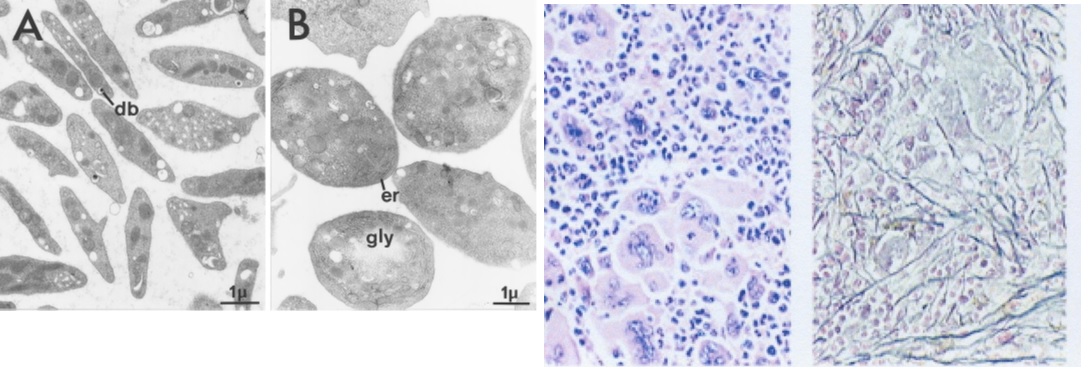
Figure 2.Normal platelets in control mice (A) and large platelets in TPOhigh mice (B). Bone marrow histology showing a significant increase of large mature megakaryocytes and increase of reticuline fibrosis grade 2 starting after 3 months of TPO treatment in TPO high mice in the study of Villeval et al in Blood 1997 [5].
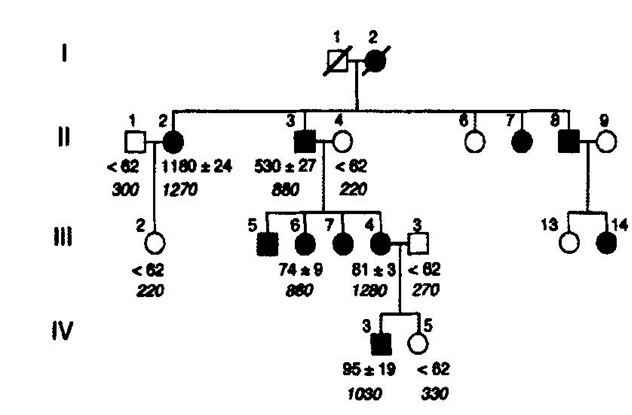
Figure 3. Pedigree of the Dutch family with hereditary essential thrombocythemia (HET). TPO serum TPO concentrations (pg/ml) from the available Dutch family members with autosomal hereditary essential thrombocythemia (HET) caused by a gain of function mutation in the TPO gene [7,8]. Filled in symbols affected individuals with germline gain of function mutation in the TPO gene.
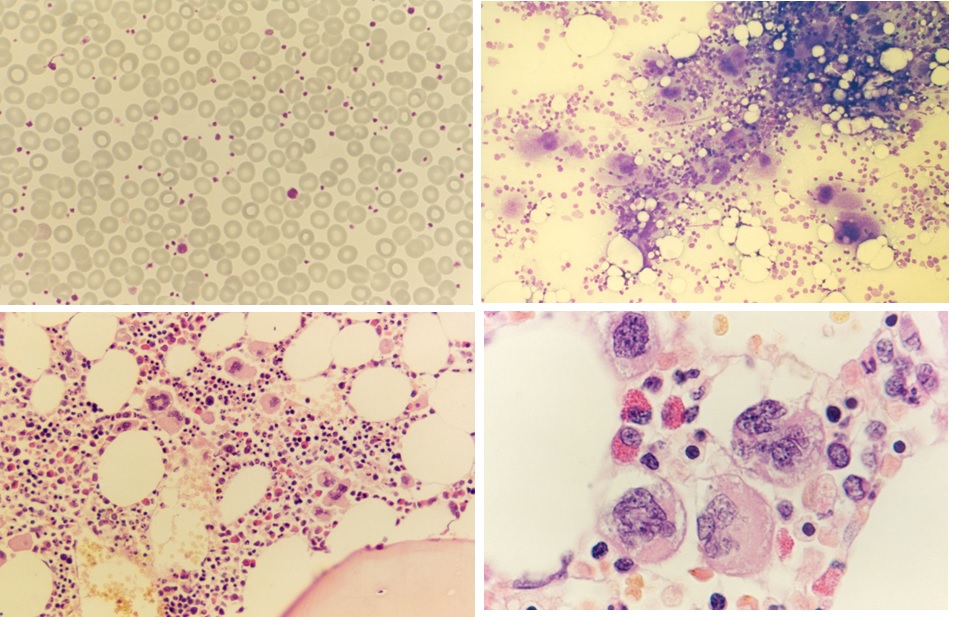
Figure 4. Peripheral blood showing some large platelets, bone marrow smear with increase of dense clustered large megakaryocytes and bone marrow histology (1986) with increase of large mature megakaryocytes in a normal cellular bone marrow and no increase of reticulin fibrosis (RF 0/1) in the proband C3 of the Dutch HET family with a gain of function mutation in the TPO gene.
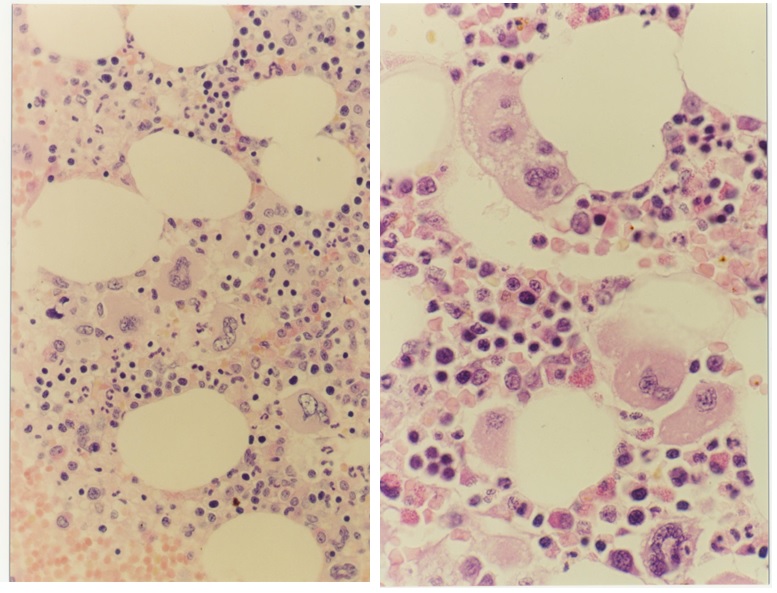
Figure 5. Bone marrow morphology (1991) showing increase and clustering of large megakaryocytes in normal cellular bone marrow and biopsy in the proband C3 of the Dutch HET family caused by a gain of function mutation in the TPO gene.
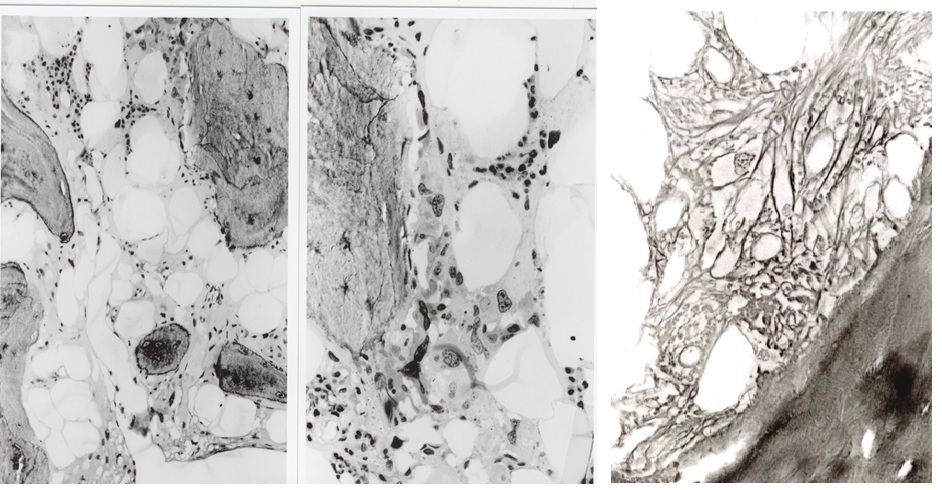
Figure 6. Bone marrow histology (1996) in the proband C3 at age 63 of the Dutch HET family caused by a gain of function mutation in the TPO gene showing a hypocellular bone marrow with the increase of reticulin fibrosis (RF 3, MF 2) and a few clustered megakaryocytes with hypolobulated nuclei.
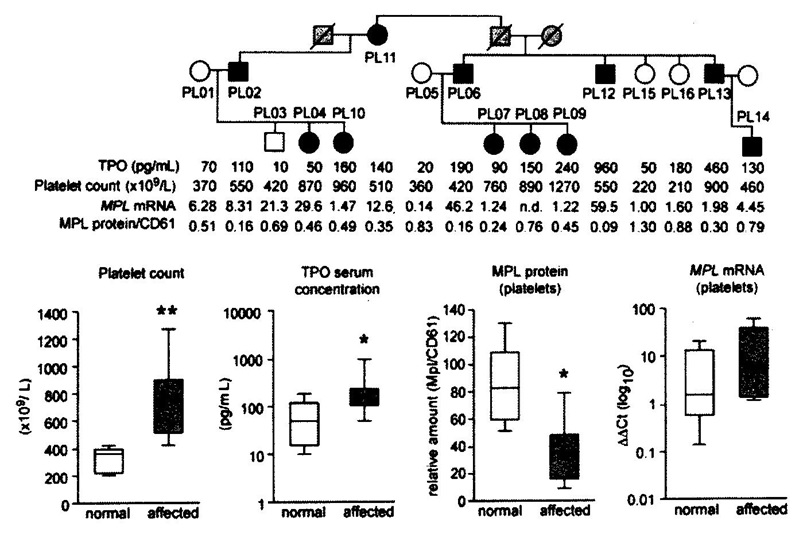
Figure 7.Upper. Pedigree of the Polish family with hereditary essential thrombocythemia (HET) due to germline gain of function mutation in the TPO gene (black) and related increased plasma TPO levels and platelet counts in affected as compared to non-affected family members [14]. Lower. Comparative analysis of platelet count, plasma TPO levels, MPL receptor membrane expression and MPL RNA messenger levels of platelets in controls and in affected members of the Polnish family with HET duet o gain of function mutation in the TPO gene [14].
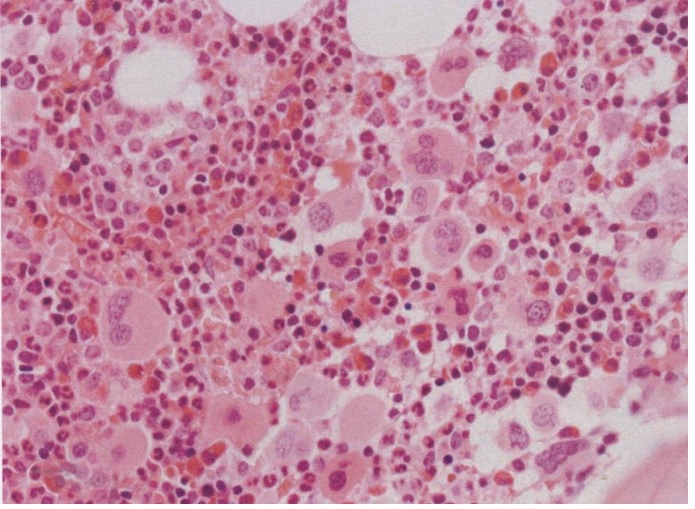
Figure 8. Bone marrow histology showed the increase and loose to dense clustering of medium-sized to large mature megakaryocytes, and normal myeloid to the erythroid ratio of bone marrow nucleated cells, no to slight increased cellularity according to age, no increase of erythropoiesis, and no increase of reticulin fibrosis (RF grade 0 to 1). As compared to controls, the clustered mature normal and medium-sized megakaryocytes were more compact with hyperlobulated nuclei [14].
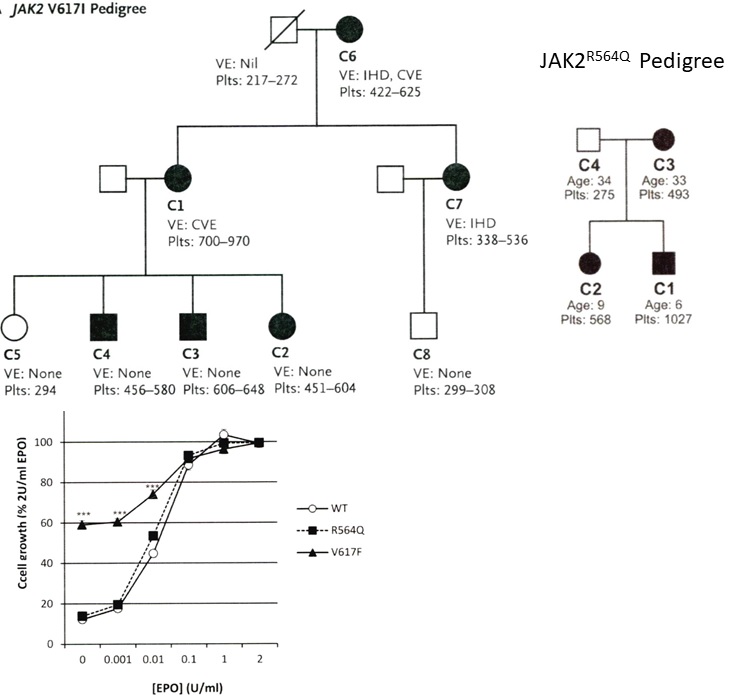
Figure 9. Upper. Pedigree of the UK family with dominant hereditary essential thrombocythemia (HET) due to germline gain of function mutation JAKV617I showing the presenting vascular events (VE) IHD = ischemic heart disease and CVE = ischemic cerebrovascular event related to increased platelet counts in the affected JAK2V617I mutated HET patients (Table 4) [24,25]. Lower. Pedigree of the UK family with dominant hereditary essential thrombocythemia (HET) due to germline gain of function mutation JAK2R564Q (Table 4) [26]. Affected JAK2R564Q mutated individuals do need EPO to induce spontaneous endogenous erythroid (EEC) colony formation in JAK2R564Q HET as compared to spontaneous EEC in acquired JAK2V617F ET [26].
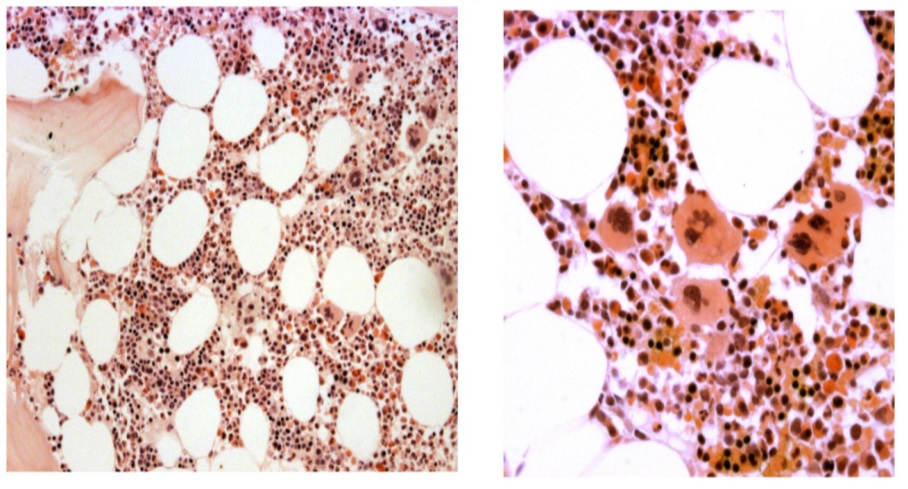
Figure 10. Upper left and right, lower left. Bone marrow histology from case C1 (left) and case C4 (right) of the family with JAK2V617I mutated HET showing a normal cellular bone marrow with increase and clustering of large megakaryocytes with some lobulation of the nuclei consistent with ET (Table 4)[24.25].
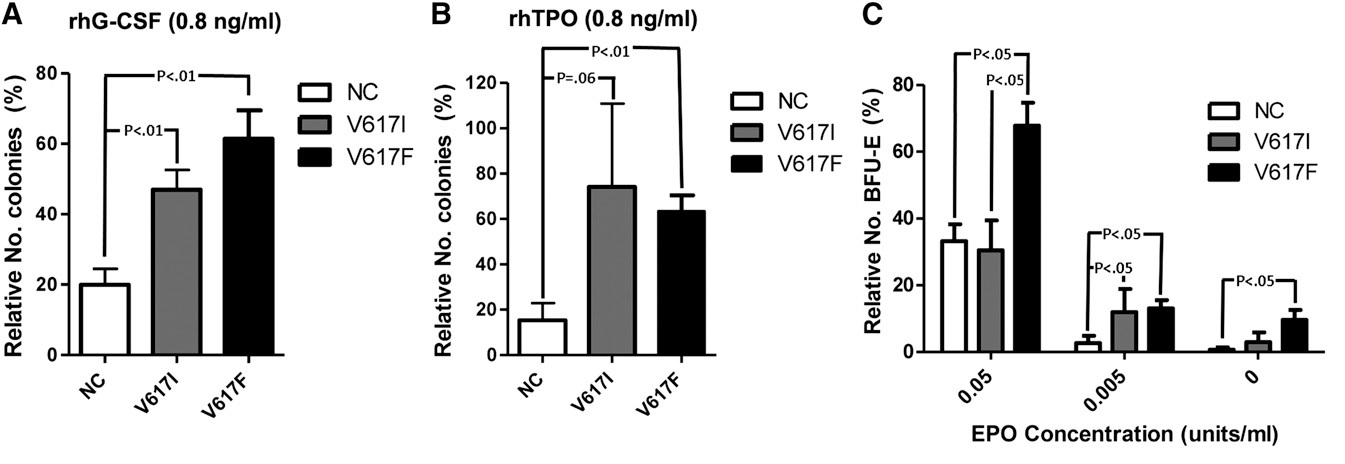
Figure 11. Response (number of colonies) of CD33+ myeloid and CD34+ hematopoietic bone marrow progenitor cells to recombinant TPO (0.8 ng/ml, left) and to EPO concentration (units/ml, right) in normal controls (NC), in congenital JAK2V617I mutated HET patients and in JAK2V617F mutated ET patients. The responses to TPO are equally increased in JAK2V671I and JAK2V617F mutated CD33 and CD34+ cells, but the response to EPO is normal in JAK2V671I and increased in JAK2V617F mutated hematopoietic progenitor cells in the study of Mead et al [25].
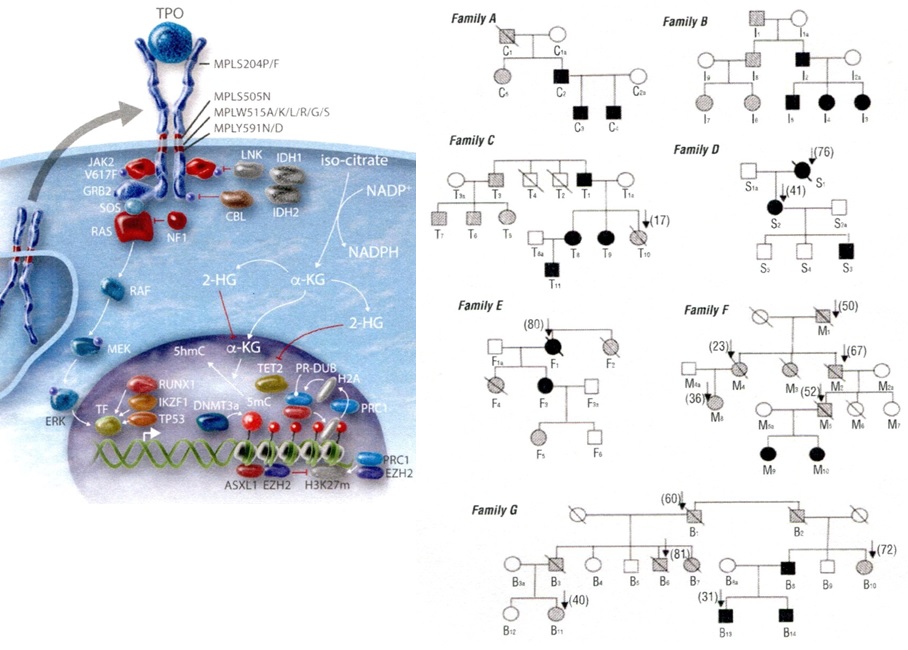
Figure 12.Lefts. Molecular etiology of dominant TPO mediated hereditary essential thrombocythemia (HET), MPLS505N mutated HET, JAK2V671I and JAK2R564Q mutated HET and MPL515 and JAK2V617F mutated acquired essential thrombocythemia (ET) according to Vainchenker & Kralovics in Blood 2017 [40]. Right. Dominant Hereditary Essential Thrombocythemia (HET) caused by germline MPLS505N mutation in 21 affected members from seven families pedigrees and the molecular etiology of congenital and acquired MPL mutated thrombocythemias without features of PV [28].
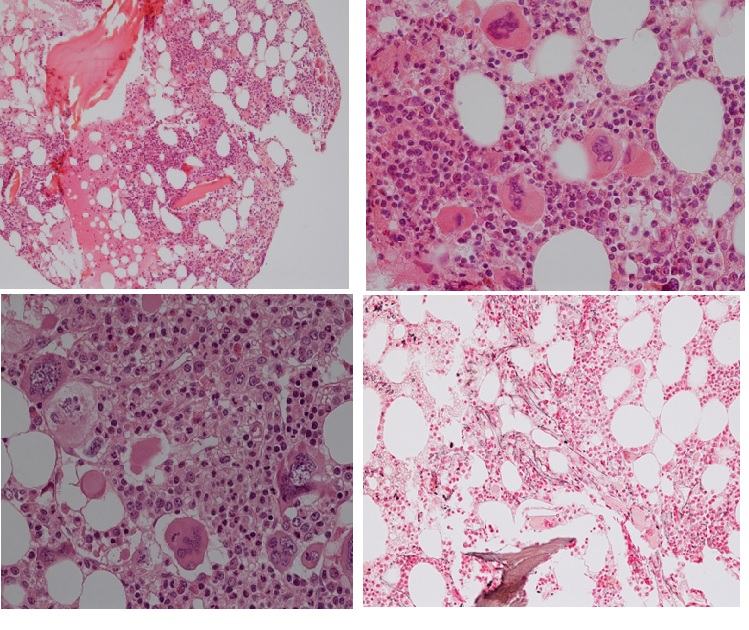
Figure 13. Bone marrow histology from a woman, age 78 at time of diagnosis of MPL515 mutated ET featured by platelet count of 1273x109/L, normal red and white blood cells, no splenomegaly and bone marrow histology showing increase and clustering of large and giant mature megakaryocytes with hyperlobulated staghorn-like in a normocellular bone marrow with increased reticulin fibrosis (RF 2).
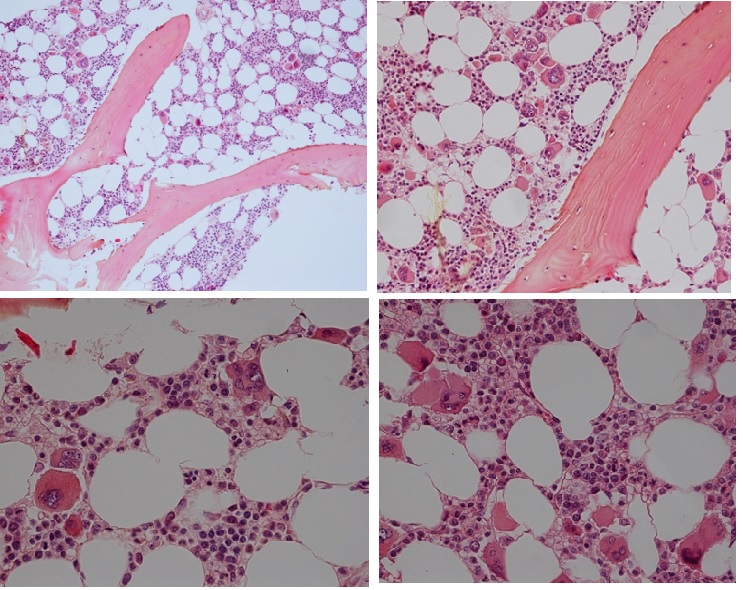
Figure 14. Bone marrow histology of CALR mutated ET in an asymptomatic man, age 39, normally red and white blood cells, no splenomegaly and bone marrow histology showing dense clusters of large immature megakaryocytes with immature bulky, cloud-like nuclei in the normocellular bone marrow and no increase of reticuline fibrosis (RF 0).
Clinical data of Dutch HET patients and non-HET subjects |
HET (%) |
Non-HET (%) |
Number of cases |
10 |
5 |
Age years, range |
11-58 |
|
Vaso-occlusive symptoms: erythromelalgia: E* |
50 |
0 |
Early E, tip paresthesia |
30 |
0 |
Red congested erythromelalgia |
10 |
0 |
E complicated by cold tip feeling-acrocyanosis/gangrene |
20 / 10 |
0/0 |
Transient ischemic attack: TIA |
30 |
0 |
Angina pectoris |
20 |
0 |
Bone marrow histology data of ten HET patient [7] |
(%) |
|
Megakaryopoiesis -increased |
100 |
|
Megakaryocyte clustering of 2-4 cells |
100 |
|
Erythropoiesis increased |
10 |
|
Reticulin-content increased grade 1 |
60 |
|
Storage iron present |
90 |
|
Chromosome abnormalities |
0 |
|
Table 1. Clinical manifestations and bone marrow histology data in the Dutch family with autosomal dominant hereditary essential thrombocythemia (Dutch HET)[7]
E*. While on low dose aspirin since 1986 all TPO mutated HET patients were free of microvascular ischemic circulation disturbances at stable platelet counts above 400x109/L and to around 1000x109/L was associated with increased plasma TPO levels during the life long follow up without the need of myelosuppressive treatment [7,8].
Case HET |
Age |
Hb |
RBC |
WBC |
Platelets |
TPO HET associated symptoms |
|||
PL Family |
Years |
g/dL |
x1012/L |
x109/L |
x109/L |
|
|
|
|
PL11 |
84 |
15.0 |
5.0 |
10.6 |
550 |
Acrocyanosis gangrene foot |
|
||
PL12 |
59 M |
15.6 |
5.5 |
5.3 |
550 - 560 |
not available |
|
|
|
PL13 |
58 M |
15.0 |
5.0 |
7.7 |
510 |
not available |
|
|
|
PL06 |
56 M |
14.5 |
5.0 |
6.5 |
408 - 410 |
None |
|
|
|
PL02 |
50 F |
13.1 |
4.5 |
5.9 |
545 - 560 |
None |
|
|
|
PL04 |
30 F |
12.3 |
4.7 |
5.9 |
595 - 1300 |
headaches, hypertension |
|
||
PL07 |
28 F |
13.2 |
4.7 |
6.1 |
760 - 960 |
TIA, miscarriage erythromelalgia |
|||
PL08 |
24 F |
13.5 |
4.7 |
7.1 |
750 - 890 |
erythromelalgia venous thrombosis |
|||
PL09 |
24 F |
12.7 |
4.1 |
6.7 |
740-1340 |
Transient ischemic attacks (TIA) |
|||
PL10 |
15 F |
14.2 |
4.1 |
10.6 |
960 |
None |
|
|
|
PL14 |
14 M |
12.3 |
4.6 |
6.2 |
460 |
None |
|
|
|
Table 3. Clinical and laboratory features in 11 affected members of the Polish (PL) family with autosomal dominant hereditary essential thrombocythemia (HET) caused by germline gain of function mutation in the TPO gene[14].
JAK2 V617I case |
C1 |
C2 |
C3 |
C4 |
C6 |
C7 |
|
Age at diagnosis |
53 |
34 |
36 |
38 |
79 |
61 |
|
Gender |
|
F |
F |
M |
M |
F |
F |
Hemoglobin g/dL |
15.7 |
14.7 |
15.6 |
14.1 |
15.4 |
14.9 |
|
Erythrocytes x 1012/L |
4.6 |
4.7 |
4.9 |
4.9 |
4.6 |
4.7 |
|
MCV fl |
|
100 |
92 |
97 |
89 |
100 |
93 |
WBC x109/L |
10.6 |
9.6 |
8.3 |
8.9 |
7.2 |
6.8 |
|
Neutrophils x109/L |
5.9 |
5.5 |
4.5 |
4.3 |
3.9 |
4.7 |
|
Platelets x 109/L |
750 |
600 |
648 |
526 |
645 |
445 |
|
EPO IU/L |
|
nt |
9.1 |
5.9 |
10.6 |
nt |
nt |
TPO pg/mL |
nt |
99 |
113 |
79 |
nt |
nt |
|
Ferritin ug/L |
nt |
127 |
146 |
290 |
nt |
nt |
|
V617I allelic level |
|
|
|
|
|
|
|
MCN % |
|
51 |
52 |
49 |
50 |
51 |
51 |
CD66+ % |
|
51 |
51 |
50 |
50 |
nt |
nt |
Table 4. Laboratory characteristics of six affected members of dominant JAK2V617I positive Hereditary Essential Thrombocythemia (HET). Platelet counts in 6 affected family members ranged from 445 to 750x109/L [24,25].
Category HET vs ET |
Number patients |
Platelet x109/L Range |
Plasma TPO pg/mL |
EEC |
Serum EPO |
||||
Dutch TPO HET |
10 |
880 |
1280 |
increased |
neg |
normal |
|||
Polish TOP HET |
11 |
701 |
1340 |
increased |
neg |
normal |
|||
JAK2 V617I HET |
6 |
445 |
750 |
normal |
neg |
normal |
|||
JAK2 V617F ET |
6 |
425 |
814 |
normal |
Pos |
decreased |
|||
MPL S505N HET |
|
|
|
normal |
neg |
normal |
|||
No splenomegaly |
9 |
627 |
1726 |
|
|
|
|||
Splenomegaly (S) |
11 |
317 |
1060 |
|
|
|
|||
MPL W515L/KET |
|
|
|
|
|
|
|||
WHO ET |
|
23 |
380 |
1500 |
|
neg |
normal |
||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
Table 5. Characteristic laboratory findings in TPO, JAK2V617I and MPLS505Nmutated hereditary essential thrombocythemia (HET) versus JAK2V617F and MPL515 mutated acquired essential thrombocythemia (ET).