Diabetes-Related Cardiomyopathy, Ischemic Heart Disease, and Heart Failure are all Associated with Changes in Mitochondrial Fatty Acid Oxidation
Received Date: December 26, 2023 Accepted Date: January 26, 2024 Published Date: January 29, 2024
doi: 10.17303/jpdm.2024.7.101
Citation: Hari Prasad Sonwani, Aakanksha Sinha (2024) Diabetes-Related Cardiomyopathy, Ischemic Heart Disease, and Heart Failure are all Associated with Changes in Mitochondrial Fatty Acid Oxidation. J Pharmacol Drug Metab 7: 1-14
Abstract
Globally, heart disease is one of the main causes of death. A decrease in cardiac efficiency and contractile dysfunction are caused by alterations in the mitochondrial energy metabolism of the heart in various heart disease types, such as diabetic cardiomyopathies, ischemic heart disease, and heart failure. Particular alterations in metabolism comprise a proportional rise in heart fatty acid oxidation rates and a disconnection between glycolysis and glucose oxidation. Heart failure can cause a general impairment in mitochondrial oxidative metabolism, whereas ischemic heart disease causes an impairment in energy production because of a restricted oxygen supply. Under these two circumstances, mitochondrial glucose oxidation is subordinated to residual mitochondrial fatty acid oxidation. The ratio of cardiac fatty acid oxidation to glucose oxidation also rises in diabetes, mostly as a result of increased fatty acid oxidation and decreased glucose breakdown. According to recent research, improving cardiac performance in ischemic hearts, failing hearts, and diabetic cardiomyopathies may be achieved by therapeutically controlling cardiac energy metabolism by decreasing fatty acid oxidation and/or boosting glucose oxidation. This article examines the metabolic changes in cardiac mitochondrial energy that are present in these types of heart disease, the role that changes in mitochondrial fatty acid oxidation play in contributing to cardiac dysfunction, and the possibility of treating these types of heart disease by focusing on fatty acid oxidation.
Keywords: Cardiomyopathies; Mitochondrial; Heart Failure
Abbreviations: ACC: acetyl CoA carboxylase; CPT-1: carnitine palmitoyl transferase 1; FABP: fatty acid binding protein; KO: knockout; CAD: long-chain acyl CoA dehydrogenase; MCAD: medium-chain acyl CoA dehydrogenase; MCD: malonyl Coa decarboxylase; PDH: pyruvate dehydrogenase; PDK4: pyruvate dehydrogenase kinase 4; PGC-1α: PPARγco-activator-1α; TAC: transverse aortic constriction; TG: triacylglycerol; TZD: thiazolidinedione
Introduction
Many types of cardiac illness are associated with changes in the metabolism of mitochondrial energy [1]. Numerous heart disease conditions, such as heart failure, ischemic heart disease, and diabetic cardiomyopathies, have been linked to mitochondrial dysfunction and decreased energy generation [1,2]. Furthermore, in certain types of cardiac disease, there may be changes in the connection between mitochondrial fatty acid oxidation and glucose oxidation. According to studies by [2], there is a rise in the fraction of fatty acids oxidized by mitochondria compared to carbohydrates oxidized due to this. Decreasing cardiac efficiency and possibly contributing to the observed decreased heart function are increases in the quantity of fatty acid oxidized by the mitochondria in relation to glucose oxidation cardiac function observed in diabetic cardiomyopathies, ischemic heart disease, and heart failure. Interestingly, there is mounting evidence that heart function can be enhanced by directly enhancing glucose oxidation or indirectly by reducing fatty acid oxidation in order to modulate cardiac energy metabolism. Among the methods employed in fundamental and applied research to attain this metabolic impact are PPARαago-nists [3,4], inhibitors of fatty acid oxidation such as trimetazidine [5,6], inhibitors of mitochondrial fatty acid uptake such as perhexiline and malonyl CoA decarboxylase (MCD) inhibitors and activators of pyruvate dehydrogenase (PDH), the rate-limiting enzyme involved in glucoseoxidation [7,8]. The processes controlling cardiac fatty acid oxidation, the function of fatty acid oxidation in cardiac diseases (diabetic cardiomyopathy, ischemic heart disease, and heart failure in particular), and the possibility of using fatty acid oxidation inhibition to treat cardiac disease are the main topics of this review.
Oxidation of cardiac fatty acids
Fatty acids and carbohydrates are the energy sources that the heart uses most frequently [1]. The majority of the cofactors required for mitochondrial oxidative phosphorylation are found in fatty acids, which are the heart's primary energy substrate. Tissue-specific fatty acid transporter proteins, CD36/FAT, and fatty acid binding protein (FABP) are among the cell membrane's fatty acid transporters that allow fatty acids to enter the cell [1]. Then, fatty acyl CoAsynthetase adds an A CoA group to the fatty acid, enabling long-chain fatty acids to enter the mitochondria. As part of this process, long-chain fatty acyl CoA is changed into an acyl carnitine by carnitine palmitoyl transferase 1 (CPT-1), which permits access into the mitochondria. The long-chain fatty acylcarnitine is subsequently transported across the inner mitochondrial membrane by carnitine translocase. The fatty long-chain After that, acyl carnitine is changed back into a fatty acyl CoA, which starts the fatty acid oxidation process. These proteins are not necessary for medium-chain fatty acids to enter the mitochondria. Every fatty acid oxidation cycle results in the production of FADH2, NADH, and acetyl CoA. The NADH and FADH2 generated by the electron transport chain are used by the tricarboxylic acid cycle, glucose oxidation, fatty acid oxidation, and glycolysis in the synthesis of ATP.
Fatty Acid Oxidation
The glucose/fatty acid cycle and malonyl CoA are two of the many variables that control By blocking CPT-1, the first protein implicated in mitochondrial long-chain fatty acid absorption, malonyl CoA controls fatty acid oxidation (McGarry et al., 1977; 1978; Paulson et al., 1984). Acetyl CoA carboxylase (ACC) and MCD are the two proteins that mainly regulate malonyl CoA and acetyl CoA levels. Acetyl CoA is carboxylate by ACC to create malonyl CoA. Malonyl CoA is changed back to acetyl CoA by MCD. Therefore, it would be anticipated that inhibiting MCD would lower CPT-1 activity, which would in turn reduce fatty acid oxidation by raising malonyl CoA levels while lowering ACC. in 1963, et al. In the heart, increasing the oxidation of fatty acids reduces the oxidation of glucose, while increasing the oxidation of glucose inhibits the oxidation of fatty acids. Through a number of processes, fatty acid oxidation reduces the metabolism of glucose. Acetyl CoA and NADH, which are generated during fatty acid oxidation, block PDH [9]. Moreover, elevated citrate levels have the ability to directly inhibit hexokinase via raising glucose-6-phosphate levels and indirectly inhibit the glycolytic enzyme phosphofructokinase 1 [1]. This additional change toward a higher proportion of cardiac energy coming from fatty acid oxidation—a less effective energy source than glucose oxidation in terms of ATP produced per O2 molecule consumed—helps explain why increased Fatty acid oxidation can be inhibited by increased glucose oxidation, as previously indicated. Acetyl CoA derived from PDH inhibits the fatty acid oxidation enzyme 3-ketoacyl CoA thiolase (Olowe and Schulz, 1980). Moreover, 3-hydroxyacyl CoA dehydrogenase, an enzyme involved in fatty acid oxidation, is inhibited by NADH generated during the oxidation of glucose (Eaton et al., 1998). Fatty acids are a less efficient energy source than glucose because when glucose is oxidized, more ATP is produced for every unit of O2 consumed. The complete oxidation of one glucose molecule results in the consumption of six O2 and the production of 31 ATP. One palmitate requires 23 O2 to oxidize, but only 105 ATP is produced. Given that just 10% of the reduction is explained by this mechanism The drop in cardiac efficiency reported in hearts with elevated rates of fatty acid oxidation is due to a combination of other mechanisms, although elevated fatty acid oxidation can cause a 30% decrease in cardiac efficiency [1] Uncoupling proteins and enhanced triacylglycerol (TG) cycling are two of these mechanisms [1].
Oxidation of Fatty Acids in Heart Disease
Depending on the kind of heart illness, there are different changes to the way the heart uses its energy. Although the effects of these various forms of cardiac disease on fatty acid oxidation are unclear, generally speaking, fatty acid oxidation rates are either raised or increased in relation to glucose oxidation rates. This is thought to be responsible for at least some of the heart function impairment since fatty acid consumption for ATP synthesis reduces cardiac efficiency. The next section discusses the changes in fatty acid oxidation and the underlying causes of these changes in heart failure, ischemic heart disease, and diabetic cardiomyopathy.
Heart Attack
The decreased cardiac activity of failing hearts is thought to be caused by changes in the source of energy substrates as well as a deficiency in energy production (Figure 2). The next section discusses the function of energy metabolism in heart failure as well as common conditions such as diabetic cardiomyopathy and myocardial ischaemia that can cause heart failure. Cardiac failure is characterized by a reduction in mitochondrial oxidative metabolism, which lowers ATP levels by 30–40% and cardiac phosphocreatine levels by a significant amount [10]. The main cause of this is the emergence of mitochondrial malfunction and the corresponding decline in mitochondrial respiration. The extent to which the specific stage and underlying etiology of heart failure determine the decline in mitochondrial function [1]. Glycolysis rates are increased in an effort to make up for the decline in mitochondrial oxidative metabolism [11-13]. The metabolic alterations observed are in line with the failing heart reverting to a fetal energy metabolism, which is distinguished by heightened glycolysis and a diminished ability for mitochondrial oxidative metabolism [11-13]. This change in total metabolic rates is accompanied by alterations in expression and activity in hypertrophied hearts caused by pressure overload. PPARα (decreased), PPARα (increased), and PPARγ co-activator-1 (PGC-1)α (decreased) are examples of transcriptional proteins [14]. When taken together, this supports a rise in glycolysis and a fall in mitochondrial oxidative metabolism. Reduced fatty acid oxidation rates are linked to lower mitochondrial oxidative metabolism, but so are decreased rates of glucose and lactate oxidation [15,16]. Protons can be produced as a consequence of an increase in the disconnection between glycolysis and glucose oxidation caused by high rates of glycolysis and low rates of glucose oxidation [17]. This may set off a chain of events that compromise ionic homeostasis and cause ATP to be diverted from contractile activity to the restoration of ionic homeostasis, ultimately lowering cardiac output. Angiotensin II is frequently utilized in the clinical environment to treat heart illness. It has recently been highlighted as a potentially significant regulator of cardiac energy metabolism and function (Mori et al 1 receptor antagonists).
Angiotensin II increases the generation of reactive oxygen species, which damages the mitochondria in the cardiomyocyte [18]. Fatty acid oxidation is one aspect of mitochondrial oxidative phosphorylation that is impacted by angiotensin II. According to Pelieux et al. (2006), transgenic mice (TG1306/R1 animals) that overexpress angiotensinogen in the myocardium have decreased cardiac fatty acid oxidation rates concurrent with decreased expression of PPARα protein and fatty acid oxidation enzymes, namely medium-chain acyl CoA dehydrogenase (MCAD) and CPT-1. Angiotensin II induces down-regulation of mRNA and protein expressions of in adult rat cardiomyocyte cells. The anti-TNF-α antibody can inhibit the genes CD36, MCAD, and CPT-1 that are involved in fatty acid oxidation (Pellieux et al., 2009). These findings imply that angiotensin II influences the oxidation of fatty acids rather than glucose. On the other hand, mounting data also suggests that angiotensin II controls the oxidation of glucose (Mori et al., 2012; 2013a). According to Mori et al. (2012), angiotensin II most likely decreases glucose oxidation by upregulating the expression of pyruvate dehydrogenase kinase 4 (PDK4), which in turn causes a reduction in PDH activity and a selective decrease in the oxidation of carbohydrates. Angiotensin II-induced upregulation of PDK4 expression can also result in insulin resistance, which can lower cardiac efficiency by forcing the heart to convert from using glucose to fatty acids as an energy source (Mori et al., 2013a). These changes in angiotensin II-induced It is possible that these abnormalities in cardiac energy metabolism lead to the development of diastolic dysfunction because they occur before diastolic failure. Angiotensin II may induce diastolic dysfunction by increasing intracellular calcium levels as a result of intracellular acidosis brought on by a greater dissociation between glucose oxidation and glycolysis. Because the elimination of the ATP-consuming cytoplasmic calcium, which is required to maintain and achieve adequate relaxation, lowers cardiac efficiency. Furthermore, angiotensin II can lower ATP levels by impairing oxidative metabolism, which lowers ATP synthesis.
Reperfusion and Ischemia
Overall mitochondrial oxidative metabolism falls with ischaemia in direct proportion to the heart's decreased oxygen supply. Elevated circulating fatty acids contribute, at least in part, to the total cardiac fatty acid oxidation rates observed during reperfusion of the ischemic heart [19]. Furthermore, there is a change in the subcellular regulation of fatty acid oxidation, leading to a deregulation of this process. Among these is a reduction in malonyl CoA brought on by an ischemic episode, which is often a strong inhibitor of the uptake of mitochondrial fatty acids (Figure 1) [20]. During reperfusion, fatty acid oxidation rates rise due to elevated circulating fatty acid levels and a substantial decrease in glucose oxidation caused by a drop in malonyl CoA levels quotations (Figure 2). The Randle cycle, which posits that increased fatty acid oxidation rates restrict glucose oxidation by blocking PDH activity, has been used to explain this. The idea that fatty acid oxidation can reduce cardiac function and efficiency, particularly during ischaemia/reperfusion, is supported by the rise in circulating fatty acids and heart fatty acid oxidation during these conditions (Figure 2) [2]. When glucose oxidation is reduced during ischaemia or reperfusion, there may be a greater uncoupling of glycolysis from glucose oxidation. This can lead to an increase in lactate and proton generation, which can lower cardiac efficiency and compromise cardiac function [2]. Lactate buildup and proton accumulation reduce cardiac efficiency. This is due to the mechanisms that are entailed in eliminating If ATP is not used to maintain ionic equilibrium, these protons can result in a buildup of intracellular calcium and sodium [1,17]. The protons are transported out of the cell by the Na+/H+ exchanger, which reduces the increasing transarcolemmal proton gradient. On the other hand, sodium is simultaneously transferred into the cell by this exchanger. After that, the Na+/Ca2+ exchanger reverses its function and calcium enters the cell as sodium intracellular levels stabilize. The calcium is then transferred out of the cell by ATP, preserving the proper intracellular calcium levels. The process of using ATP to primary Maintaining ionic homeostasis lowers heart rate effectiveness.
Diabetic Heart Disease
Ventricular dysfunction in individuals with diabetes mellitus, regardless of the presence of other coronary artery disorders, is known as diabetic cardiomyopathy. One of the most prevalent and expensive chronic illnesses, diabetes mellitus, is also a major contributor to heart disease. Recent epidemiological data indicate that diabetes mellitus affects more than 25% of Americans over 65. One of the most serious side effects and leading cause of death for those with diabetes mellitus is cardiovascular disease. Actually, diabetes mellitus increases the chance of developing a cardiac attack (McDonald & Associates, 2000). Diabetes-related cardiomyopathies are thought to be influenced by changes in the heart's mitochondrial energy metabolism [21]. For example, increased rates of fatty acid oxidation are found in the hearts of humans or animals suffering from obesity or diabetes mellitus [1]. As a result, there is a significant drop in the rates at which glucose is oxidized (Figure 2), and the diabetic or obese heart's primary energy source becomes mitochondrial fatty acid oxidation. According to Buchanan et al. (2005), these changes in cardiac energy metabolism occur before glucose intolerance and ventricular hypertrophy. It is becoming evident that abnormalities in energy metabolism and cardiac function seen in diabetic cardiomyopathy are caused by excessively high fatty acid oxidation rates, even though the role of energy metabolism is probably much more complex [21]. Both the buildup of fatty acids in the heart and high rates of fatty acid oxidation may be the mechanisms by which an excess of fatty acids leads to the development of diabetic cardiomyopathy. Heart failure is likely exacerbated by cardiac lipotoxicity, which is frequently linked to diabetes mellitus. Fatty acids have the ability to be converted into fatty acid intermediates, such as ceramides and diacylglycerol (DAG), which may worsen cardiac function by increasing insulin resistance in the heart [22,23]. According to [23], DAG is thought to be the main lipid intermediary in the heart that contributes to insulin resistance. Although TG is not thought to directly cause myocardial insulin resistance, a number of studies have also demonstrated a buildup of myocardial TG in obesity and diabetes mellitus [23]. It is still unknown what processes lead to the build-up of these lipid intermediates. Two possible explanations have been put out to account for the build-up of intramyocellular lipid: (i) compromised fatty acid oxidation, and (ii) excess fatty acid supply. There is inconsistent evidence to support the theory that intramyocardial lipid buildup is caused by poor mitochondrial fatty acid oxidation [1]. Myocardial fatty acid oxidation rates are high in diabetics, according to the majority of studies conducted in humans (Rijzewijk et al., 2009; Peterson et al., 2012) and animals [21]. Fatty acid oxidation rates are likewise higher with obesity (Peterson et al., 2004; Axelsen et al., 2012). That's comparable.
The higher than normal levels of circulating fatty acids associated with diabetes are a more plausible reason for the buildup of lipids in the diabetic myocardium [21]. Many factors, such as high insulin levels, are involved. to these elevated quantities of free fatty acids in the blood. Insulin promotes the synthesis of TG and inhibits lipolysis in adipose tissue. Elevated amounts of circulating free fatty acids are caused by increased lipolysis in adipose tissue and hydrolysis of TG in the context of insulin resistance, such as Type 2 diabetes.
Another mechanism that contributes to the increased fatty acid supply to the heart in diabetics is cellular fatty acid absorption. cardiac disease. The fatty acid transporters CD36 and FABP are persistently relocated from the cytosol to the cell membrane in conjunction with cardiac insulin resistance (Luiken et al., 2001; Coort et al., 2004; Carley et al., 2007). Chronic rise in fatty acid absorption caused by persistent translocation of CD36 or FABP fatty acid transporters may be responsible for the enhanced myocardial fatty acid oxidation seen in diabetic hearts (Chabowski et al., 2005). db/db mice have decreased myocardial glucose oxidation rates and increased cardiac fatty acid oxidation as early as 4 weeks of age (Buchanan et al., 2005). Studies on the genetics of diabetic cardiomyopathy suggest that fatty acid uptake-related proteins may be implicated in the disease's progression. For instance, decreasing Heart insulin resistance brought on by a high-fat diet is less severe when FABP expression is present (Shearer et al., 2008). Crucially, heart function was unaffected negatively by this partial suppression of FABP3 expression (Shearer et al., 2008). Furthermore, lipotoxic cardiomyopathy is caused by the overexpression of additional fatty acid uptake-related proteins, such as lipoprotein lipase and long-chain acyl-CoA synthetase [24].
These changes in the metabolism of cardiac fatty acids are likewise mediated by the PPARs. The PPAR family's endogenous ligands are fatty acids. The expression of genes controlled by the PPARs, which include enzymes involved in fatty acid oxidation, is upregulated by fatty acids and their derivatives. Heart mitochondrial fatty acid oxidation rates are elevated when PPARα increases the expression of CD36, CPT-1, MCD, and long-chain acyl CoA dehydrogenase (LCAD) [25]. Insulin resistance and diabetes mellitus are associated with increased expression of PPARα, indicating a potential involvement of this transcription factor in the increased transport and oxidation of fatty acids seen in diabetic hearts [26]. Moreover, the PPARα overexpressing mice's phenotype is similar to Type 2 elevated fatty acid oxidation rates are associated with diabetes mellitus [26]. However, in db/db mice aged 15–18 weeks, additional studies have demonstrated that while the expression of PPARα-regulated genes, like MCAD, LCAD, and mCPT-1, is increased, the expression of PPARα itself is not enhanced [26]. The absence of variation in PPARα expression could indicate that modifications in heart metabolism in db/db animals are not dependent on PPARα, or it could imply that PPARα activity is increased without regard to protein expression. In db/db mice, there is an increase in the production of PGC-1, the PPARα co-activator, which ultimately results in an increase in PPARα activity (Carley and Severson, 2005). PPARα also alters the expression of PDK4, which decreases the rate of glucose oxidation by phosphorylating PDH. Through the Randle cycle, PPARα activation lowers glucose oxidation rates, which in turn contributes to the high rates of fatty acid oxidation in the mitochondria. This could be the mechanism underlying the altered metabolism of energy in diabetic hearts. The elevated myocardial fatty acid oxidation seen in diabetes mellitus is mostly due to this excess of fatty acids and the PPARs that are activated as a result. All things considered, the evidence points to an excess of fatty acids as the cause of the heart lip toxicity seen in diabetic cardiomyopathy. Lipid intermediates may build up if the amount of fatty acids exceeds the rate at which they are oxidized. But this wouldn't be directly caused by decreased fatty acid oxidation because, when diabetes is present, fatty acid oxidation rates often rise rather than fall. It's crucial to remember that cardiac lip toxicity may also be a factor in other illnesses if there is a persistent increase in circulating fatty acids and compromised heart function.
Treating heart disease by focusing on fatty acid oxidation
Treatment for heart failure, ischemic heart disease, and diabetic cardiomyopathy has shown promise in the inhibition of mitochondrial fatty acid oxidation. There are two ways to stop fatty acid oxidation: either directly by stopping fatty acid intake into the mitochondria or by stopping mitochondrial fatty acid oxidation, or indirectly by stopping fatty acid oxidation via increasing glucose oxidation. It is advantageous to pharmacologically inhibit fatty acid oxidation with medications such as mitochondrial fatty acid oxidation inhibitors (e.g., trimetazidine) (Figure 3), CPT-1 inhibitors (e.g., perhexiline, etomoxir), or MCD inhibitors (e.g., CBM-301106). Using PPARα or PPARγ ligands to reduce the heart's circulating supply of fatty acids is another method of limiting fatty acid oxidation (Figure 3). We won't go into these medications, but it's vital to note that increasing glucose oxidation is another way to block fatty acid oxidation, which in turn inhibits fatty acid oxidation (Figure 3). Since it is not directly blocking ATP-producing pathways, this should also be helpful in cases of severe heart failure. Directly inhibiting fatty acid oxidation may lower failing heart function and ATP levels, which are already lowered in severe heart failure. The idea that part of the etiology of decreased cardiac performance is elevated fatty acid oxidation rates, as seen in settings like reperfusion after ischaemia, is supported by the fact that lowering fatty acid oxidation can improve cardiac function. Inhibiting fatty acid oxidation may be used to treat cardiac disease. Trimetazidine is one medication that targets mitochondrial fatty acid oxidation enzymes specifically. According to [5,6], trimetazidine lowers rates of glycolysis and/or promotes glucose oxidation, which lowers proton levels and improves the function of failing hearts. Not all research, nevertheless, has shown that trimetazide-treated hearts had lower rates of fatty acid oxidation [27]. One likely contributing factor is that Saeedi et al. used 1.2 mM palmitate in their perfusate, whereas Kantor et al. used 0.4 mM palmitate in their studies, reporting a decrease in fatty acid oxidation rates [27]. Due to According to Lipachuk et al. (2003), trimetazidine is a reversible competitive inhibitor of 3-ketoacyl CoA thiolase. Trimetazidine inhibition can be overcome by high amounts of this enzyme's sub-stratum.
Additionally, MCD inhibitors seem to hold promise in the management of cardiac conditions. Improved insulin sensitivity, reduced fatty acid oxidation, and enhanced glucose oxidation are the results of MCD inhibition [7]. Repression According to Lipachuk et al. (2003), trimetazidine is a reversible competitive inhibitor of 3-ketoacyl CoA thiolase. Trimetazidine inhibition can be overcome by high amounts of this enzyme's sub-stratum.
Additionally, MCD inhibitors seem to hold promise in the management of cardiac conditions. Improved insulin sensitivity, reduced fatty acid oxidation, and enhanced glucose oxidation are the results of MCD inhibition [7]. Repression of MCD results in increased malonyl CoA levels, which block CPT-1 and hence lower rates of fatty acid oxidation. Although the effects of MCD inhibition on cardiac energy metabolism imply that MCD inhibition would be helpful in the situation of heart failure, the effects of MCD inhibition on heart failure have not yet been investigated. Targeting PPARs is another way to treat heart disease. Fib rides and thiazolidinediones are two pharmacological families that target the PPAR transcription factor family (TZD). It is TZDs that activate PPARγ. According to [28], TZDs have a number of advantageous effects, such as lowering circulating levels of TG and fatty acids and increasing myocardial glucose oxidation, which should improve cardiac efficiency. But heart function could suffer from TZDs. Exacerbated heart failure has been documented in diabetic patients receiving TZD treatment [29]. Furthermore, the Prospective Pioglitazone Clinical Trial in Macrovascular Events (PROactive) Study (Dormandy et al.) found that the incidence of heart failure was higher in diabetic patients receiving TZDs due to a variety of factors, including TZD stimulation of vasodi-lation, which raised peripheral oedema [29]. PPARα activity is increased by fibrates. It is thought that the beneficial effects of these medications on the heart result from a decrease in myocardial fatty acid oxidation brought on by a decrease in the amount of circulating fatty acids [30]. Treatment with PPARα agonists has conflicting benefits. It has been stated that they shield the heart from damage caused by reperfusion and ischemia [31]. Fibrines did not lower coronary heart disease mortality in the FIELD research, but they did prove advantageous in the Helinski Heart research and the Va-HIT trial [4]. Increasing PPARδ activity is an additional effective treatment option for heart disease. Numerous genes, including those involved in fatty acid oxidation, are expressed more frequently when PPARδ is present [25]. It has been found that PPARδ increases myocardial glucose oxidation rates (Burkart et al., 2007) and prevents cardio-myocyte hypertrophy (Planavila et al., 2005; Pellieux et al., 2009). It seems counterintuitive that PPAR agonists have a positive impact on heart disease because one would think that enhance cardiac fatty acid oxidation by upregulating the expression of the fatty acid oxidation-related proteins, which are typically thought to impair heart function (as previously discussed). However, PPAR agonists reduce the amount of circulating fatty acids by enhancing extracardiac fatty acid oxidation, which lowers cardiac fatty acid oxidation rates (Lopaschuk et al., 2010). As was previously indicated, increased glycolysis and the uncoupling of glycolysis from glucose oxidation may be the mechanism by which elevated fatty acid oxidation worsens heart performance. Despite higher rates of fatty acid oxidation, the quantity of glycolysis uncoupled from glucose oxidation is not raised in ACC2 KO mice, suggesting that their hearts may operate normally. Additionally, there is proof that decreasing cardiac fatty acid oxidation is harmful to a failing heart. For instance, even though markers of fatty acid oxidation should decline, lowering the quantity of circulating fatty acids in heart failure further impairs cardiac function (Tuunanen et al. Although the evidence available to date points to a significant role for fatty acid oxidation in the development and management of heart illness, this relationship is not entirely clear-cut. For instance, current research using the ACC2 knockout (KO) mice does not clearly corroborate the theory that heart function is diminished by fatty acid oxidation (Kolwicz). Although the evidence available to date points to a significant role for fatty acid oxidation in the development and management of heart illness, this relationship is not entirely clear-cut. For instance, current research using ACC2 knockout (KO) mice does not clearly corroborate the idea that fatty acid.
Endpoint
The metabolism of cardiac energy, particularly the oxidation of fatty acids, seems to play a significant role in the pathogenesis of heart disease. Although elevated rates of mitochondrial fatty acid oxidation may not necessarily correspond with cardiac illness, they seem to play a significant role in many cases in the observed compromised heart function. Heart disease may more frequently be caused by the increasing uncoupling of glycolysis from glucose oxidation as a result of fatty acid oxidation-induced suppression of glucose oxidation. The uncoupling of glycolysis from glucose oxidation is actually increased in severe end-stage heart failure, where fatty acid oxidation is actually decreased. It makes sense that medications that promote glucose oxidation will enhance heart health. However, inhibiting fatty acid oxidation is a potential target for the therapy for heart conditions. However, depending on the particular cardiac illness, efforts to treat it by blocking fatty acid oxidation will need to be carefully addressed in order to avoid aggravating an already serious condition where any decrease in ATP levels is harmful.
- Lopaschuk GD, Tsang H (1987) Metabolism of palmitate in isolated working hearts from spontaneously diabetic ‘BB’ Wistar rats. Circ Res 61: 853-8.
- Liu Q, Docherty JC, Rendell JCT, Clanachan AS, Lopaschuk GD (2002) High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol 39: 718-25.
- Yue TL, Bao W, Jucker BM, Gu JL, Romanic AM, Brown PJ et al. (2003) Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation 108: 2393–9.
- Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR et al. (2005) Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366: 1849-61.
- Fragasso G, Palloshi A, Puccetti P, Silipigni C, Rossodivita A, Pala M et al. (2006a) A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol 48: 992–8.
- Fragasso G, Perseghin G, De Cobelli F, Esposito A, Palloshi A, Lattuada G et al. (2006b) Effects of metabolic modulation by trimetazidine on left ventricular function and phosphocreatine/adenosine triphosphate ratio in patients with heart failure. Eur Heart J 27: 942–8.
- Dyck JR, Cheng JF, Stanley WC, Barr R, Chandler MP, Brown S et al. (2004) Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res 94: e78–84.
- Cheng JF, Huang Y, Penuliar R, Nishimoto M, Liu L, Arrhenius T et al. (2006) Discovery of potent and orally available malonyl-CoA decarboxylase inhibitors as cardioprotective agents. J Med Chem 49: 4055–8.
- Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD (2011). Targeting fatty acid and carbohydrate oxidation a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta 1813: 1333–50.
- Conway MA, Allis J, Ouwerkerk R, Niioka T, Rajagopalan B, Radda GK (1991) Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet 338: 973–6.
- Lei B, Lionetti V, Young ME, Chandler MP, d’Agostino C, Kang E et al. (2004) Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J Mol Cell Cardiol 36: 567–76.
- Degens H, de Brouwer KF, Gilde AJ, Lindhout M, Willemsen PH, Janssen BJ et al. (2006). Cardiac fatty acid metabolism is preserved in the compensated hypertrophic rat heart. Basic Res Cardiol 101: 17–26.
- Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y et al. (2010). Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail 3: 420–30
- Keller A, Rouzeau JD, Farhadian F, Wisnewsky C, Marotte F, Lamande N et al. (1995). Differential expression of alpha- and beta-enolase genes during rat heart development and hypertrophy. Am J Physiol Heart Circ Physiol 269: H1843–51.
- Zhabyeyev P, Gandhi M, Mori J, Basu R, Kassiri Z, Clanachan et al. (2013) Pressure-overload-induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc Res 97: 676–85.
- Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M et al. (2013) Cardiac insulin resistance and decreased mitochondrial energy production precede the development of systolic heart failure following pressure overload hypertrophy. Circ Heart Fail 6: 1039–48.
- Dennis SC, Gevers W, Opie LH (1991) Protons in ischemia: where do they come from; where do they go to? J Mol Cell Cardiol 23: 1077–86.
- Dai DF, Johnson SC, Villarin JJ, Chin MT, NievesCintron M, Chen T et al. (2011) Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res
- Folmes CD, Sowah D, Clanachan AS, Lopaschuk GD (2009) High rates of residual fatty acid oxidation during mild ischemia decrease cardiac work and efficiency. J Mol Cell Cardiol 47: 142–8.
- Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD (1995) High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-- CoA levels due to an increase in 5’-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem 270: 17513–20.
- How OJ, Larsen TS, Hafstad AD, Khalid A, Myhre ES, Murray AJ et al. (2007) Rosiglitazone treatment improves cardiac efficiency in hearts from diabetic mice. Arch Physiol Biochem 113: 211–20.
- Zhang L, Keung W, Samokhvalov V, Wang W, Lopaschuk GD (2010) Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim Biophys Acta 1801: 1–22.
- Zhang L, Ussher JR, Oka T, Cadete VJ, Wagg C, Lopaschuk GD (2011) Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res 89: 148–56.
- Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P et al. (2001) A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 107: 813–22.
- Yang Q, Li Y (2007) Roles of PPARs on regulating myocardial energy and lipid homeostasis. J Mol Med 85: 697–706.
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A et al. (2002) The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 109: 121–30.
- Kantor PF, Lucien A, Kozak R, Lopaschuk GD (2000) The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res 86: 580–8.
- Zhu P, Lu L, Xu Y, Schwartz GG (2000) Troglitazone improves recovery of left ventricular function after regional ischemia in pigs. Circulation 101: 1165–71.
- Lindenfeld J, Masoudi FA (2007) Fluid retention with thiazolidinediones: does the mechanism influence the outcome? J Am Coll Cardiol 49: 1705–7.
- Cook WS, Yeldandi AV, Rao MS, Hashimoto T, Reddy JK (2000) Less extrahepatic induction of fatty acid beta-oxidation enzymes by PPAR alpha. Biochem Biophys Res Commun 278: 250–7.
- Yue T-L, Bao W, Gu J-L, Cui J, Tao L, Ma X-L et al. (2005) Rosiglitazone treatment in zucker diabetic fatty rats is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes 54: 554–62.
- Coort SL, Hasselbaink DM, Koonen DP, Willems J, Coumans WA, Chabowski A et al. (2004) Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese zucker rats. Diabetes 53: 1655–63.
- Daniels A, van Bilsen M, Janssen BJ, Brouns AE, Cleutjens JP, Roemen TH et al. (2010). Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiol (Oxf) 200: 11–22
- Donnelly R, Emslie-Smith AM, Gardner ID, Morris AD (2000) ABC of arterial and venous disease: vascular complications of diabetes. BMJ 320: 1062–6.
- Dormandy JA, Charbonnel B, Eckland DJA, Erdmann E Massi-Benedetti M, Moules IK et al. (2005) Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 366: 1279–89.
- Eaton S, Middleton B, Bartlett K (1998) Control of mitochondrial beta-oxidation: sensitivity of the trifunctional protein to [NAD+]/[NADH] and [acetyl-CoA]/[CoA]. Biochim Biophys Acta 1429: 230–8.
- Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C et al. (2006) Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol 47: 598–604.
- Kolwicz SC, Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R (2012) Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res 111: 728–38.
- Lopaschuk GD, Barr R, Thomas PD, Dyck JR (2003) Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res 93: e33–7.
- Tuunanen H, Engblom E, Naum A, Nagren K, Hesse B, Airaksinen KE et al. (2006) Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 114: 2130–7.
- Ussher JR, Folmes CD, Keung W, Fillmore N, Jaswal JS, Cadete VJ et al. (2012a) Inhibition of serine palmitoyl transferase I reduces cardiac ceramide levels and increases glycolysis rates following diet-induced insulin resistance. PLoS ONE 7: e37703.
- Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T et al. (2012b) Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res 94: 359–69.
- Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y et al. (2003) Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest 111: 419–26.
FIGURE 1
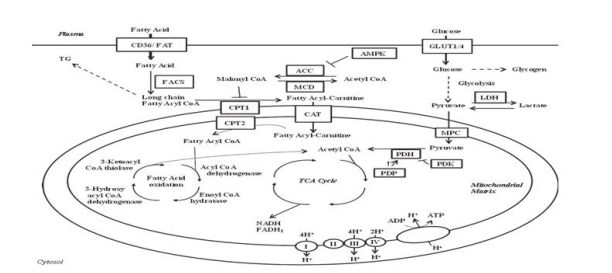
Figure 1: Overview of fatty acid and glucose oxidation in the heart
ACC, acetyl CoA carboxylase; AMPK, AMP- activated protein kinase; CPT, carnitine palmitoyl transferase; CAT, carnitine translocase; FACS, fatty acyl CoA synthetase; FAT, fatty acid transporter; GLUT, glucose transporter; LDH, lactate dehydrogenase; MCD, malonyl CoA decarboxylase; MPC, mitochondrial pyruvate carrier; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PDP, pyruvate dehydrogenase phosphatase; TCA, tricarboxylic acid; TG, triacylglycerol
FIGURE 2
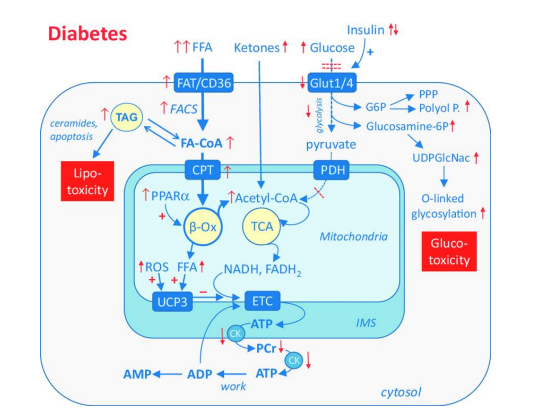
Figure 2: Cardiac metabolic alterations in diabetes. In diabetes, strongly increased free fatty acid activate peroxisome proliferator-activated receptor a (PPARa), which up-regulates expression of genes involved in fatty acid (FA) oxidation. Increased FA oxidation shuts down glucose uptake and oxidation (insulin resistance), thereby blunts metabolic flexibility. Excessive FA are stored as triacylglycerol (TAG), which can mediate lipotoxicity. FA and reactive oxygen species (ROS) activate uncoupling protein 3 (UCP3), which makes ATP production less efficient
FIGURE 3
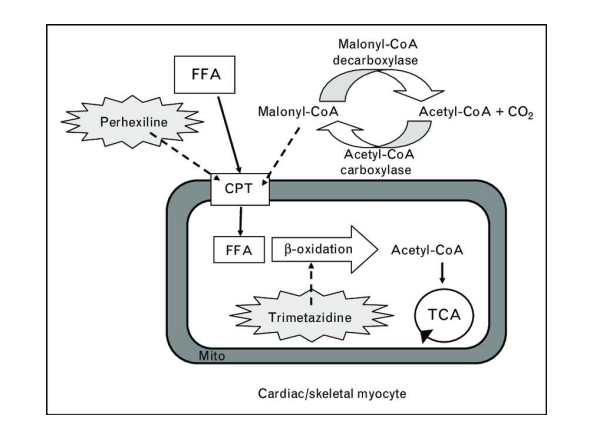
Figure 3: Mechanisms of cardioprotection of fatty acid oxidation inhibitors
FIGURE 4
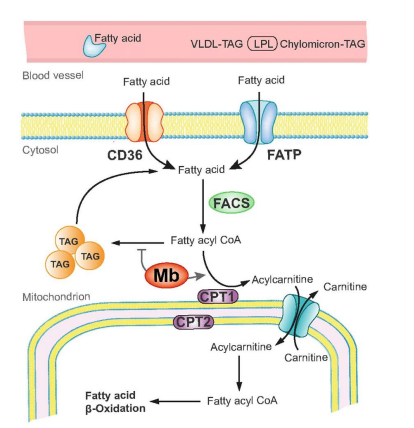
Figure 4: Potential role of myoglobin in cardiac fatty acid metabolism. The heart is supplied with fatty acids (FA) derived from plasma FAs bound to albumin or incorporated in very-low-density lipoproteins (VLDL) triacylglycerols (TAG) or chylomicrons. The uptake occurs via diffusion or CD36/FATP transporters. Within the cytosol FAs are converted to FA acyl CoA by fatty acyl coA synthetases (FACS). The majority of FA acyl CoA is then channeled to carnitine palmitoyltransferase (CPT) system mediated by myoglobin (Mb). Converted to acylcarnitine by CPT1 FAs are translocated into the mitochondrial matrix, where they are converted back to fatty acyl CoA by CPT 2 to enter the β oxidation cycle. A minor portion of FA acyl CoA is converted to TAG under the control of Mb.
Figures at a glance