Metabolic Changes and Immune Escape of IL-34 in p53 Mutated Head and Neck Cancers
Received Date: February 05, 2026 Accepted Date: March 05, 2026 Published Date: March 11, 2026
doi:10.17303/jpdm.2026.9.101
Citation: Shuchun Lin, Xiaohua Hu, Yunhai Gao, Zu-Chian Chiang, Jizhen Lin (2026) Metabolic Changes and Immune Escape of IL-34 in p53 Mutated Head and Neck Cancers. J Pharmacol Drug Metab 9: 1-12
Abstract
TP53 gene mutation is one of the most common genetic changes in head and neck squamous cell carcinoma (HNSCC), with an incidence rate of 33% -58%, especially in HPV negative cases. TP53 gene mutation not only leads to the inactivation of the anti-cancer function of the original p53 protein, but also enables it to acquire carcinogenic activity (mutation acquiring new functions), driving tumor development, treatment resistance, and poor prognosis. In p53 protein mutated HNSCC, high expression of IL-34 is directly associated with poor prognosis and has become a key factor in predicting poor survival. IL-34 forms an immunosuppressive microenvironment through CD36 mediated lipid metabolism reprogramming, thereby inhibiting CD8+ T cell function and promoting immune escape. This review analyzes and summarizes the various roles of IL-34 in HNSCC, and discusses its relationship with TP53 gene mutations, promotion of cancer stem cells CSCs, and significance of clinical immune therapies.
Keywords: Metabolic changes; Immune escape; Mutant p53; IL-34; Head and Neck Cancers
Introduction
Head and neck squamous cell carcinoma (HNSCC) is a common and aggressive cancer that results in significant morbidity and mortality worldwide, posing a serious threat to public health. Genetic alterations play a critical role in the initiation and progression of HNSCC, with TP53 gene mutations being one of the most prevalent changes, occurring in 33% to 58% of cases, especially in human papillomavirus (HPV)-negative tumors [1, 2]. The TP53 gene encodes the p53 protein, an essential tumor suppressor that regulates cell cycle arrest, apoptosis, DNA repair, and senescence to maintain genomic stability [3]. However, mutations in TP53 not only disrupt the normal tumor-suppressive functions of wild-type p53 but also give the mutant p53 protein new gain-of-function (GOF) properties [2, 4]. These properties promote tumor proliferation, metastasis, treatment resistance, and immune evasion, ultimately leading to poorer clinical outcomes.
In recent years, interleukin-34 (IL-34) has been recognized as a key cytokine involved in the development of various cancers, including HNSCC [5, 6]. IL-34 shares the colony-stimulating factor 1 receptor (CSF1R) with macrophage colony-stimulating factor (M-CSF) but has distinct structural and functional characteristics [7]. High levels of IL-34 expression in HNSCC, particularly in cases with TP53 mutations, have been closely associated with poor prognosis, including reduced overall survival (OS) and disease-free survival (DFS), as well as an increased risk of recurrence and metastasis [1, 8]. Clinical data from the Cancer Genome Atlas (TCGA)-HNSCC cohort further confirm that IL-34 serves as an independent prognostic marker, unaffected by TNM staging and HPV status [1]. Additionally, tumors that express IL-34 are often resistant to immunotherapies, such as programmed death-1 (PD-1) inhibitors, which limit the effectiveness of current treatment strategies [8, 9].
The mechanism connecting TP53 mutations to the overexpression of IL-34 and tumor progression has been receiving increasing attention. Under normal physiological conditions, wild-type p53 functions as a transcription factor that directly binds to a conserved sequence in the promoter region of the IL-34 gene, thereby inhibiting its transcriptional activity [10]. However, mutations in TP53 (such as missense mutations and deletions) lead to the inactivation of p53, which relieves this transcriptional repression and results in uncontrolled secretion of IL-34 [1, 11]. This dysregulated production of IL-34 creates a critical interaction with mutant p53, reshaping the immunosuppressive tumor microenvironment TME and driving the generation and maintenance of cancer stem cells (CSCs) [1, 12]. CSCs are a small subpopulation of tumor cells characterized by their self-renewal, differentiation, and tumorigenic capabilities, making them significant contributors to treatment resistance, metastasis, and tumor recurrence [12, 13]. IL-34 directly promotes the enrichment of CSCs by upregulating stem cell markers (e.g., CD44, CD133) and activating stemness-related transcription factors (e.g., Nanog, Oct4, Sox2) [14, 15]. Furthermore, IL-34 plays a role in reprogramming lipid metabolism within the TME through the CD36 receptor, which is a key regulator of fatty acid uptake and oxidation [16, 17]. IL-34 increases the expression of CD36 on tumor-associated macrophages (TAMs) via the p38 mitogen-activated protein kinase (MAPK) pathway. This enhancement promotes the uptake of free fatty acids (FFAs) and supports fatty acid oxidation (FAO) [16, 18]. Such metabolic reprogramming leads to the polarization of TAMs toward the M2 phenotype (characterized as foam-like M2 TAMs), which secrete immunosuppressive factors like interleukin-10 (IL-10) and transforming growth factor-β (TGF-β), thereby inhibiting the function of CD8⁺ T cells [19, 20]. Additionally, M2 TAMs compete with CD8+ T cells for nutrients in the TME, resulting in T cell energy depletion and functional exhaustion [21, 22]. Moreover, CD36-mediated lipid peroxidation can trigger ferroptosis in CD8⁺ T cells, further impairing their anti-tumor activity [23, 24]. IL-34 may also increase the expression of immune checkpoint molecules, such as programmed death-ligand 1 (PD-L1), on both CSCs and M2 TAMs via the signal transducer and activator of transcription 3 (STAT3) pathway [2, 9]. This forms a dual inhibitory network that exacerbates immune evasion.
The IL-34/mutant p53 axis plays a critical role in the progression of HNSCC, immune evasion, and resistance to treatment. Targeting this axis shows great potential for improving outcomes in patients with p53-mutated HNSCC. Preclinical studies have indicated that combining IL-34/CSF1R blockade with PD-1 inhibitors can synergistically enhance anti-tumor immunity, resulting in high rates of complete remission and the induction of long-term immune memory [1, 9]. Several targeted agents, such as IL-34 monoclonal antibodies (e.g., AM-001) and CSF1R inhibitors (e.g., pimicotinib/ABSK021), have begun clinical trials, opening new avenues for precision therapy. Additionally, targeting downstream molecules like CD36 or key signaling pathways (e.g., PI3K/Akt, MAPK/ERK) could potentially reverse metabolic reprogramming and restore anti-tumor immune responses [17, 25].
This review aims to systematically summarize the various roles of IL-34 in p53-mutated HNSCC, highlighting its molecular mechanisms related to metabolic reprogramming, immune evasion, and CSC regulation. We will also discuss the clinical significance of targeting the IL-34/mutant p53 axis and its potential to overcome treatment resistance, offering insights for the development of novel combination therapeutic strategies for this challenging subset of HNSCC.
Molecular basis of p53 mutation driving IL-34 secretion
The reason why IL-34 is highly valued in HNSCC is that its high expression is closely related to tumor occurrence, development, and prognosis [5, 7, 26-28]. IL-34 activates the CSF1R receptor, promotes polarization of TAMs towards the M2 phenotype, forms an immunosuppressive microenvironment, and subsequently inhibits CD8+ T cell function [11], accelerating tumor growth and meta stasis [6, 29]. During the treatment process, IL-34 positive tumors are prone to developing resistance to immunotherapy (such as PD-1 inhibitors) [8], leading to treatment failure. Table 1 data shows that patients with high IL-34 expression have a worse prognosis and a higher risk of recurrence. Clinical doctors need to pay attention to the status of IL-34 in order to optimize treatment strategies, such as combination therapy targeting IL-34, which is expected to improve patient survival outcomes.
In recent years, research done by Wei Haiming's team at the University of Science and Technology of China has revealed that the inactivation of p53 protein and abnormal expression of IL-34 form an axis [1, 28]. This axis is not only the core of reshaping the immunosuppressive TME, but also a key engine driving the generation and maintenance of CSCs. IL-34 directly promotes the formation and amplification of CSCs with high tumorigenicity, self-renewal ability, and immune escape characteristics through its mediated complex network, providing a new perspective for understanding the malignant biological behavior of p53 mutated HNSCC.
In HNSCC with TP53 gene mutations [4, 27, 28], the transcriptional inhibition of IL-34 by p53 protein is relieved, which is the key reason for the elevation of cytokine IL-34 [11]. Under normal physiological conditions, p53 acts as a transcription factor and can directly bind to the conserved sequence of the IL-34 gene promoter region (such as RRRCWWGYYY), inhibiting its transcription [10]. TP53 gene mutations (such as missense mutations and deletions) lead to the inactivation of p53 protein function, resulting in the loss of transcriptional repression ability towards IL-34. This situation is caused by uncontrolled cell growth, high expression of IL-34, and expression of various cancer stem cell markers.
After p53 is inactivated due to mutation, cancer cells become the main secreting cells of IL-34, promoting the production of CSCs [1, 4, 27]. Single cell sequencing data showed that CSCs highly expressed IL-34 in p53 inactivated tumors, while other cell types expressed extremely low levels [29, 32]. Therefore, p53 mutation → inactivation of transcriptional repression on IL-34 → massive secretion of IL-34 → production of CSCs is a key chain in promoting cancer, typically due to the inactivation of the gene TP53 that controls cell growth, proliferation, and metastasis.
IL-34 promotes tumor immune escape through lipid metabolism reprogramming
After secreting IL-34, CSCs mainly reshape the TME by binding to the receptor CSF1R, forming an ecosystem conducive to the growth, survival, and expansion of CSCs [4, 12]. The main characteristic of this ecosystem is immune suppression, manifested by macrophage polarization into a subtype filled with fat droplets, namely M2 TAMs, a typical product of the immune suppressive TME [19, 29]. The activation of downstream PI3K/Akt and JAK2/STAT3 pathways by IL-34/CSF1R signaling drives reverse metabolic changes in macrophages. M2 TAMs highly express markers CD206 and CD163, and secrete immunosuppressive factors such as IL-10 and TGF-β, which inhibit the toxicity of CD8+ T cells towards cancerous cells [33]. This is the M2 TAMs foam, thus promoting tumor immune escape. The core mechanism of this process involves the synergistic effect of IL-34 and CD36 receptors, which reshape the energy metabolism network of macrophages by regulating fatty acid uptake and oxidative metabolism, ultimately leading to the cessation of local immune response in tumors (Figure 1).
IL-34 significantly increases the expression of CD36 on macrophage surfaces by activating the p38 MAPK signaling pathway [16]. CD36, as a long-chain fatty acid transporter, can efficiently recognize and bind to free fatty acids (FFAs) enriched in the tumor microenvironment, mediating their transmembrane uptake [34]. Once fatty acids enter the cell, the mitochondrial beta oxidation pathway is activated, and fatty acids are converted to acylcarnitine by carnitine palmitoyltransferase (CPT) catalysis. They then enter the mitochondrial matrix and undergo continuous dehydrogenation, hydration, and thiolysis reactions, ultimately producing acetyl CoA. Acetyl CoA enters the tricarboxylic acid cycle (TCA) to produce NADH and FADH2, providing electrons for the electron transport chain and driving ATP synthesis [35]. This process not only provides energy for macrophages, but also leads to excessive accumulation of lipid metabolic intermediates (such as triglycerides), forming lipid droplets in cells, giving macrophages typical foam like shape.
However, this lipid metabolism reprogramming has a profound impact on CD8+ T cells in the tumor microenvironment [18, 36]. M2 TAMs directly inhibit T cell proliferation and activation by secreting immunosuppressive factors such as IL-10 and TGF–β [20]. At the same time, limited nutrients in the tumor microenvironment are preferentially taken up by M2 TAMs, leading to a shortage of energy metabolism in T cells [21]. T cells rely on glycolysis for energy supply, but in a lipid rich environment, their mitochondrial FAO ability is inhibited, leading to insufficient ATP synthesis [22]. In addition, the lipid metabolites of M2 TAMs, such as reactive oxygen species (ROS), further damage the mitochondrial function of T cells, exacerbating their energy crisis [37]. Ultimately, T cells lose their cytotoxic function due to energy depletion and are unable to effectively kill tumor cells.
The IL-34/CD36 axis promotes tumor immune escape through a dual mechanism
on the one hand, it directly induces macrophages to polarize towards M2 subtype, forming an immunosuppressive microenvironment; On the other hand, by competitively ingesting lipids, T cells are deprived of their energy supply, leading to their functional paralysis [1]. This process reveals the core mechanism of immune cell metabolic interactions in the tumor microenvironment, providing a theoretical basis for targeted CD36 or IL-34 immunotherapy.
The above changes also promote lipid metabolism reprogramming, specifically manifested in CD36 mediated fatty acid oxidation [18]. IL-34 upregulates the CD36 receptor on the surface of TAMs, enhancing their uptake of long-chain fatty acids and FAO metabolism [11]. This led to the accumulation of lipid droplets in TAMs, forming a typical foam like M2 macrophage morphology. This is the main reason for the formation of metabolic changes to promote the foam of macrophages [37]. The CD36 driven FAO metabolism consumes a large amount of lipids in the TME, causing CD8+ T cells to lose their function due to energy depletion and unable to effectively attack tumor cells [17].
In addition, in the TME, CD36 mediated lipid peroxidation and ferroptosis are key mechanisms that weaken the tumorigenic ability of CD8+ T cells [4, 23]. High expression of CD36 promotes T cell uptake of excessive fatty acids, especially unsaturated fatty acids, leading to the accumulation of lipid peroxides and triggering ferroptosis [17]. This process is accompanied by the accumulation of iron ions and an increase in reactive oxygen species (ROS) levels, further exacerbating cell damage [38]. Ferroptosis not only reduces the secretion of cytotoxic factors by CD8+ T cells, but also decreases their proliferation and survival ability, thereby significantly weakening the anti- tumor effect [24]. Preclinical studies have shown that inhibiting CD36 or using ferroptosis inhibitors can restore T cell function and enhance the efficacy of immunotherapy [23]. Therefore, targeting the CD36 lipid peroxidation axis provides a new strategy for overcoming immunotherapy resistance.
On the other hand, IL-34 can also upregulate the expression of immune checkpoints through the STAT3 pathway, such as PD-L1 expression on CSCs and M2 TAMs [29], thus forming a dual inhibition, namely, dual metabolic and immune checkpoint inhibition, providing an explanation for the limited response of p53 mutated tumors to PD-1 monoclonal antibody therapy [9, 27]. Research data shows that patients with TP53 gene mutations have limited survival extension after receiving PD-1 immune checkpoint inhibitor treatment [39], and some patients even have a shorter survival period. These clinical phenomena require further exploration.
TP53 gene mutations can reduce the immunogenicity of tumor cells, decrease the production of new antigens, and make it difficult for the immune system to recognize tumor cells [2]. At the same time, it promotes the infiltration of immune suppressive cells (such as Treg and MDSC) and upregulates immune checkpoint molecules such as PD-L1, forming a local immunosuppressive TME, making it difficult for PD-1 antibodies to function [40]. TP53 gene mutations shape the immunosuppressive microenvironment, promote tumor evolution, and suppress immune cell function, collectively leading to a decrease in the efficacy of PD-1 inhibitors [2] and even a shortened survival period in some cases. Future treatments may require a combination of targeting the TP53 pathway, chemotherapy, or radiotherapy to overcome this challenge.
The role of IL-34 in directly inducing and maintaining head and neck CSCs
IL-34 is not only a reshaping agent of the immune microenvironment, but also a direct catalyst for the production of CSCs. IL-34 directly promotes the expression of CSCs markers, for example, p53 mutations promote the production and enrichment of CSCs by releasing inhibition of stem cell markers such as CD44 and CD133 [13, 41]. IL-34 may indirectly or directly upregulate these dry markers through its receptor CSF1R or other signaling pathways. In HNSCC, CD44 has been identified as the most common CSCs marker, and CD44+ cells have strong tumorigenicity, metastatic potential, and chemoradiotherapy resistance. IL-34 participates in maintaining the CD44+ CSCs subpopulation through its signaling network.
IL34 activates the dry transcription factor the transcription factor network. In head and neck tumors, the tumor microenvironment rich in cytokines such as TGF-β and IL-6 produced by IL-34 can activate the STAT3 signaling pathway. The activation of STAT3 further promotes the expression of core dry transcription factors the transcription factor Nanog, Oct4, and Sox2 [14]. These transcription factors form a positive feedback pathway that collectively maintains the self-renewal, differentiation, and immune escape abilities of tumor stem cells. The Nanog-Stat-3 signaling axis has been confirmed to be positively correlated with the CD4+ tumor stem cell subpopulation and immune escape ability, suggesting a complete "IL-34/TGF β/Nanog/CD44/immune escape" signaling pathway.
IL-34 induced M2 TAMs physical barrier and T cell dysfunction. Transcriptome data of this process shows that IL-34+ CSCs and CD36hi TAMs co-locate at the tumor invasion edge, forming a structure with CSCs as the center and foam like M2 TAMs as the peripheral barrier [1, 11]. This physical structure envelops CSCs, isolates attacks from immune cells, and provides a safe haven for CSCs. This is an obstacle to conventional antibody immunotherapy, and further research and exploration are needed to break this barrier in the future.
Detailed mechanism of IL-34 induced tumor stem cells CSCs
IL-34 is a cytokine that functions similarly to macrophage colony-stimulating factor (M-CSF) but has a different structure, sharing the same receptor CSF1R [7]. In recent years, studies have found that IL-34 plays a key role in the tumor microenvironment with p53 protein mutations, directly or indirectly inducing, maintaining, and amplifying CSCs through complex signaling networks, thereby driving tumor progression, immune escape, and treatment resistance. The core mechanism can be summarized as the following interrelated levels.
Important trigger point: p53 inactivation and uncontrolled secretion of IL-34 under normal circumstances, the wild-type p53 protein acts as a transcription factor that directly binds to and inhibits the promoter region of the IL-34 gene [3], thereby suppressing the expression of IL-34. In the case of TP53 gene mutations, such as missense mutations and deletions, in various solid tumors such as HNSCC and hepatocellular carcinoma (HCC) lead to the inactivation of p53 protein function and the loss of transcriptional control over IL-34 [28, 32]. After the inactivation of p53 function, the CSCs secrete a large amount of IL-34, forming a self-reinforcing pro-tumor ecosystem.
The IL-34/CSF1R signaling pathway directly acts on tumor stem cells CSCs and their microenvironment
IL-34 binds to CSF1R on the surface of CSCs and TAMs, activating multiple downstream signaling pathways and forming a network that promotes the growth of CSCs. This signal network directly maintains the self-renewal and amplification of CSCs. The IL-34 signal may directly enhance the self-renewal ability, anti-apoptotic ability, and drug resistance of CSCs by activating classic survival and proliferation pathways such as PI3K/Akt and MAPK/ERK. These pathways are also crucial for maintaining stem cell characteristics. In inducing the immunosuppressive microenvironment, it indirectly supports the self-renewal of CSCs, which is the core link for IL-34 to induce and maintain CSCs.
HNSCC often exhibits polarized M2 TAMs [29]. IL-34 plays a crucial role in promoting the formation of M2 macrophages. It induces the differentiation of monocytes into macrophages with an immunosuppressive phenotype (M2) through the CSF1R pathway. This is characterized by high levels of CD163 and CD206 expression, the secretion of IL-10 and TGF-β, and low expression of IL-12. These features contribute to a typical immunosuppressive microenvironment, which is a significant reason for the ineffectiveness of PD-1 immune checkpoint therapy. Within this tumor microenvironment, PD-L1 is upregulated, facilitating immune evasion.
M2 tumor-associated macrophages (TAMs) activated by IL-34 overexpress the immune checkpoint protein PD-L1. The underlying mechanisms include: (a) a direct enhancement of PD-L1 expression in cancer stem cells (CSCs) through the STAT3 pathway, as well as upregulation of PD- L1 secretion by TAMs and other cells in the microenvironment via the PI3K/Akt and MAPK/ERK signaling pathways. PD-L1 binds to PD-1 on T cells, inhibiting their activation and killing function, particularly in CD8⁺ T cells, which creates an "immune-exempt" environment for CSCs; (b) metabolic reprogramming that hijacks CD8+ T cells. IL-34 increases the expression of the CD36 receptor on macrophages, enhancing their uptake and oxidative metabolism of FAO. This led to the metabolic reprogramming of TAMs, which turned into foam like phenotype, and consumed a large amount of lipid and other energy substances in the microenvironment, resulting in no energy available for CD8+ T cells, which led to functional exhaustion and further weakened anti-tumor immunity.
Establishing "CSCs TAMs" physical barriers and ecology
In tumors with p53 protein mutations, CSCs secreting IL-34 (high expression of EpCAM, CD47 and other biomarkers) are closely associated spatially with CD36h1 M2 TAMs recruited and polarized by them, especially forming a physical barrier structure at the forefront of tumor invasion. This structure envelops CSCs, protecting them from immune cell attacks and maintaining them in a microenvironment rich in immunosuppressive factors, hypoxia, and specific metabolites, greatly promoting the survival and continuous expansion of CSCs.
Changes in key molecular markers
The mutation of p53 protein and activation of IL-34 signaling pathway lead to upregulation of characteristic markers of CSCs. Firstly, CD44 upregulation mediates cell migration and adhesion, promoting cancer cell metastasis [15]. Secondly, upregulation of CD133 (Prominin-1) further enhances the self-renewal ability of stem cells [13]. Secondly, the elevation of aldehyde dehydrogenase (ALDH) is closely related to its high activity and resistance to radiotherapy and chemotherapy [42]. In addition, Bmi-1 upregulation enhances the self-renewal ability of CSCs [13, 43]. Finally, an important receptor of IL-34, Syndecan-1 (SDC-1), is upregulated and may also be involved in fine-tuning and interactions of the signaling network [44].
In summary, the induction of tumor stem cells CSCs by IL-34 is a multi-step, networked process. P53 mutation → increase of IL-34 secretion → activation of CSF-1R → direct maintenance of CSCs stemness+induction of M2 TAMs polarization → upregulation of PD-L1 mediated immune escape + CD36 mediated metabolic hijacking → formation of immunosuppressive microenvironment and physical barrier → ultimately leading to CSCs amplification, tumor progression, and immunotherapy resistance [1, 11, 45]. Interventions targeting this pathway, especially in combination with immune checkpoint inhibitors, provide a highly promising new direction for the treatment of p53 mutant tumors.
Clinical significance and prospects of targeted therapy
The clarification of the IL-34/p53 axis offers a novel strategy for targeted treatment of HNSCC with p53 mutations. This approach aims to improve the effectiveness of combination immunotherapy for tumors. Preclinical studies have demonstrated that PD-1 monoclonal antibodies, when used alone, have limited effectiveness in treating tumors with p53 mutations [28]. However, the combined blockade of IL-34 signaling (using IL-34 antibodies or CSF1R inhibitors) and PD-1 inhibitors can produce significant synergistic effects [1]. In animal models of p53 mutant tumors, this combination therapy has even achieved a complete remission rate of up to 90% (subcutaneous injection) and can induce long-term immune memory. In the research of targeting IL-34 transformation, antibodies targeting IL-34 (such as AM-001) and CSF1R inhibitors (such as pimicotinib/ABSK021) have entered the clinical trial stage.
In preclinical models of HNSCC, IL-34 antibodies can significantly inhibit tumor growth, reduce M2 macrophage infiltration, and restore T cell function. The combination of PD-1/LAG-3 dual immunotherapy is expected to provide a new option for PD-1 resistant patients. Targeting the IL-34-CD36 axis [1, 11] not only reverses immune suppression, but also has the potential to overcome chemotherapy and radiotherapy resistance mediated by CSCs by reshaping the microenvironment and interfering with dry signals (such as the Nanog axis [45]) in overcoming CSCs related drug resistance.
The IL-34 induced CSCs and their immunosuppressive microenvironment are important factors leading to resistance to PD-1/PD-L1 inhibitors (with a response rate of less than 20% in patients with p53 mutations). Therefore, targeting the IL-34 pathway has become a new strategy to overcome drug resistance. Firstly, a treatment regimen that directly targets IL-34, using IL-34 monoclonal antibodies, can reduce tumor size by 70% in preclinical p53 mutant liver cancer models. Combined with anti-PD-1 antibodies, it can achieve 100% complete remission. In the treatment targeting downstream pathways, CSF-1R inhibitors (such as Pexidartinib, Pimicotinib/ABSK021) are used. Blocking the common receptor of IL-34 and M-CSF can reverse the M2 polarization phenotype of TAMs, and shows synergistic efficacy when combined with PD-1 inhibitors [1, 29].
Metabolic reprogramming in tumor tissue is one of the key factors leading to the failure of cancer immune response. In the TME, cancer cells utilize metabolic hijacking mechanisms to take up lipids through fatty acid transporters such as CD36, leading to impaired immune cell function [1]. CD36 is highly expressed in tumor infiltrating regulatory T cells (Tregs) and TAMs, promoting lipid accumulation, making TAMs present a foam like phenotype, and enhancing the immunosuppressive activity of Tregs, thereby inhibiting the anti-tumor function of CD8+ T cells. In addition, CD36 mediated lipid peroxidation and ferroptosis further weaken the killing ability of T cells. Inhibitors targeting CD36 can block this metabolic hijacking, reduce foam like M2 TAMs, and restore the effector function of T cells. Preclinical studies have shown that CD36 monotherapy or in combination with chemotherapy and immunotherapy (such as PD-1 inhibitors) can significantly enhance anti-tumor immune response [1]. Additionally, combining PD-1 inhibitors and antiangiogenic agents also offers a promising strategy to enhance immunity for solid tumor therapy [46]. Currently, related combination therapies are being explored clinically, providing new strategies for overcoming immunotherapy resistance.
In terms of CD36 inhibitors, they block metabolic hijacking and restore T cell function. Single drug can reduce foam like M2 TAMs, which is under clinical exploration in combination with chemotherapy or immunotherapy. In terms of joint pathway inhibition, the combination of specific inhibitors (such as aprixib and trametinib) targeting the PI3K/Akt and MAPK/ERK pathways simultaneously activated by IL-34 may enhance the anti-tumor effect [25].
Conclusion
The mutation of p53 results in the loss of transcriptional repression of IL-34, leading to high levels of IL-34 production. This increase in IL-34 drives the generation of CSCs, which in turn secrete large amounts of IL-34, creating a vicious cycle. CSCs release IL-34, which reprograms TAMs to form an immunosuppressive and metabolically supportive microenvironment. This environment not only helps maintain, amplify, and protect CSCs but also contributes to tumor progression, metastasis, and resistance to treatment. IL-34 acts as a link between p53-mutated CSCs and is a critical hub molecule involved in immune evasion and metabolic reprogramming. Consequently, targeting IL-34 and its downstream signaling pathways, such as CSF1R and CD36, offers a strategy to reverse immune suppression. This approach presents a promising new direction for addressing the challenges posed by p53 mutations in the treatment of head and neck cancer.
Future research should focus on clarifying the specific regulatory mechanisms by which IL-34 influences the signaling pathways involved and accelerates the clinical development of related targeted therapies, providing hope for patients with poor prognoses.
Author contributions
Lin S, Hu X, and Gao Y reviewed the literature and drafted the manuscript. Chiang ZC and Lin J revised and approved the manuscript.
Funding
Supported by Natural Science Foundation of Fujian Province (grant number: 2022J01246) and Double high- level medical institution construction fund of Fujian Province (grant number: Fujian Health Medical Policy [2021] No. 76).
Conflicts of Interest
The authors declare no potential conflicts of interest.
- Nian Z, et al. (2024) Interleukin-34-orchestrated tumor-associated macrophage reprogramming is required for tumor immune escape driven by p53 inactivation. Immunity. 57: 2344-261 e7.
- Wang, C., et al., TP53 Mutation-Mediated Immune Evasion in Cancer: Mechanisms and Therapeutic Implications. Cancers (Basel), 2024. 16(17).
- Hernandez Borrero, L.J. and W.S. El-Deiry, Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim Biophys Acta Rev Cancer, 2021. 1876(1): p. 188556.
- Chen, X., et al., Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death Dis, 2022. 13(11): p. 974.
- Ge, Y., M. Huang, and Y.M. Yao, Immunomodulation of Interleukin-34 and its Potential Significance as a Disease Biomarker and Therapeutic Target. Int J Biol Sci, 2019. 15(9): p. 1835-1845.
- Igarashi, Y. and K.I. Seino, Role of IL-34 in Tumors and Its Application to Regulate Inflammation. Cancer Sci, 2025. 116(5): p. 1164-1170.
- Otsuka, R., H. Wada, and K.I. Seino, IL-34, the rationale for its expression in physiological and pathological conditions. Semin Immunol, 2021. 54: p. 101517.
- Monteleone, G., et al., Targeted Therapy of Interleukin-34 as a Promising Approach to Overcome Cancer Therapy Resistance. Cancers (Basel), 2023. 15(3).
- Alshaebi, F., et al., Interleukin-34 and immune checkpoint inhibitors: Unified weapons against cancer. Front Oncol, 2023. 13: p. 1099696.
- Brazda, V. and M. Fojta, The Rich World of p53 DNA Binding Targets: The Role of DNA Structure. Int J Mol Sci, 2019. 20(22).
- Gyorgypal, A. and R.M. Anthony, TAM-ing the beast with IL-34 blockade. Sci Immunol, 2024. 9(101): p. eadu0981.
- Zhong, H., et al., Tumor microenvironment as niche constructed by cancer stem cells: Breaking the ecosystem to combat cancer. J Adv Res, 2025. 71: p. 279-296.
- Lai, J., et al., Id1 and NF-kappaB promote the generation of CD133+ and BMI-1+ keratinocytes and the growth of xenograft tumors in mice. Int J Oncol, 2014. 44(5): p. 1481-9.
- Hu, R., Q. Han, and J. Zhang, STAT3: A key signaling molecule for converting cold to hot tumors. Cancer Lett, 2020. 489: p. 29-40.
- Senbanjo, L.T. and M.A. Chellaiah, CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front Cell Dev Biol, 2017. 5: p. 18.
- Liu, Q., et al., IL-34 promotes foam cell formation by enhancing CD36 expression through p38 MAPK pathway. Sci Rep, 2018. 8(1): p. 17347.
- Zhou, X., et al., CD36: The Bridge between Lipids and Tumors. Molecules, 2024. 29(2).
- Jin, H.R., et al., Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. J Hematol Oncol, 2023. 16(1): p. 103.
- Zhang, W., et al., Macrophage polarization in the tumor microenvironment: Emerging roles and therapeutic potentials. Biomed Pharmacother, 2024. 177: p. 116930.
- Chen, J., et al., Role of elevation of glycolysis in tumor-associated macrophages in glioblastoma immune evasion and therapeutic implications. PNAS Nexus, 2026. 5(1): p. pgaf396.
- Lobel, G.P., Y. Jiang, and M.C. Simon, Tumor microenvironmental nutrients, cellular responses, and cancer. Cell Chem Biol, 2023. 30(9): p. 1015-1032.
- Ma, S., et al., Cellular metabolism regulates the differentiation and function of T-cell subsets. Cell Mol Immunol, 2024. 21(5): p. 419-435.
- Ma, X., et al., CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab, 2021. 33(5): p. 1001-1012 e5.
- Liang, Y., et al., Ferroptosis: CD8(+) T cells' blade to destroy tumor cells or poison for self-destruction. Cell Death Discov, 2025. 11(1): p. 128.
- Chen, C.P., et al., Synergistic effects of the combination of trametinib and alpelisib in anaplastic thyroid cancer with BRAF and PI3KCA co-mutations. Heliyon, 2024. 10(7): p. e29055.
- de Bakker, T., et al., Restoring p53 Function in Head and Neck Squamous Cell Carcinoma to Improve Treatments. Front Oncol, 2021. 11: p. 799993.
- Tsai, C.C., et al., Immune Evasion in Head and Neck Squamous Cell Carcinoma: Roles of Cancer-Associated Fibroblasts, Immune Checkpoints, and TP53 Mutations in the Tumor Microenvironment. Cancers (Basel), 2025. 17(15).
- Cai, B.H., et al., Strategies for Anticancer Treatment in p53-Mutated Head and Neck Squamous Cell Carcinoma. Biomedicines, 2025. 13(5).
- Wang, J., et al., Combined single-cell RNA-seq and bulk RNA-seq construction of M2 TAMs signature for predicting HNSCC prognosis and immunotherapy. Front Immunol, 2025. 16: p. 1620931.
- Ju, Y., et al., Genomic Landscape of Head and Neck Squamous Cell Carcinoma Across Different Anatomic Sites in Chinese Population. Front Genet, 2021. 12: p. 680699.
- Perez Sayans, M., et al., Comprehensive Genomic Review of TCGA Head and Neck Squamous Cell Carcinomas (HNSCC). J Clin Med, 2019. 8(11).
- Wang, G., et al., Single-cell transcriptome sequencing reveals spatial distribution of IL34(+) cancer-associated fibroblasts in hepatocellular carcinoma tumor microenvironment. NPJ Precis Oncol, 2023. 7(1): p. 133.
- Xu, J., et al., Dual roles and therapeutic targeting of tumor-associated macrophages in tumor microenvironments. Signal Transduct Target Ther, 2025. 10(1): p. 268.
- Chen, Y., et al., CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J Exp Med, 2022. 219(6).
- Wang, Y., et al., Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J Biol Chem, 2019. 294(33): p. 12380-12391.
- Mao,Y.X., W.J. Xia, and P. Jiang, metabolites as signalling molecules in the tumor immune microenvironment. Nature Reviews Immunology, 2026.
- Teng, Y., et al., Targeting reactive oxygen species and fat acid oxidation for the modulation of tumor-associated macrophages: a narrative review. Front Immunol, 2023. 14: p. 1224443.
- Abdukarimov, N., K. Kokabi, and J. Kunz, Ferroptosis and Iron Homeostasis: Molecular Mechanisms and Neurodegenerative Disease Implications. Antioxidants (Basel), 2025. 14(5).
- Bai, B., et al., The clinical features and prognostic implications of the co-mutated TP53 gene in advanced non-small cell lung cancer. Clin Transl Oncol, 2024. 26(12): p. 3236-3245.
- Tie, Y., et al., Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol, 2022. 15(1): p. 61.
- Cao, L., et al., CD44a functions as a regulator of p53 signaling, apoptosis and autophagy in the antibacterial immune response. Commun Biol, 2022. 5(1): p. 889.
- Gao, J., et al., Aldehyde Dehydrogenase 2 as a Therapeutic Target in Oxidative Stress-Related Diseases: Post-Translational Modifications Deserve More Attention. Int J Mol Sci, 2022. 23(5).
- Liu, S., et al., Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res, 2006. 66(12): p. 6063-71.
- Meyer, A., et al., Dysregulation of IL-34 ligation to SDC-1 mitigates collagen-induced arthritis. Cell Mol Immunol, 2022. 19(9): p. 1070-1072.
- Noh, K.H., et al., Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J Clin Invest, 2012. 122(11): p. 4077-93.
- Liu Q., et al., Strengthening effect of thalidomide combined with anti-PD1 antibody on enhancing immunity for lung cancer therapy. Curr Pharm Biotechnol, 2025. 26(17), 1-11.
FIGURE 1
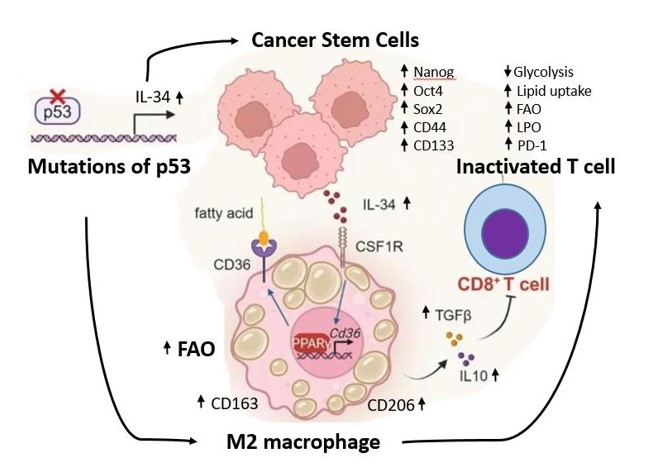
Figure 1: The p53 mutations lost its suppression on IL-34 and induced the production of CSCs. CSCs secreted a large amount of IL-34, thus contributing to the formation of M2 macrophages (foam like M2 TAMs). M2 TAMs secreted IL-10 and TGF and then inactivated CD8+ T cells.
Tables at a glance
Figures at a glance