A Novel Intratumor Immunotherapy for Prostate Cancer Targets the A1 Adenosine Receptor
Received Date: March 20, 2023 Accepted Date: April 19, 2023 Published Date: April 22, 2023
doi: 10.17303/jurh.2023.2.101
Citation: Constance N Wilson, Yongping Chen (2023) A Novel Intratumor Immunotherapy for Prostate Cancer Targets the A1 Adenosine Receptor
Abstract
The American Cancer Society estimates 268,490 new cases of prostate cancer in 2022. Moreover, the ACS estimates that 34,500 men will die from prostate cancer in 2022. In in vivo studies in a xenograft model of PC3 human prostate carcinoma in Athymic nude mice, an intratumor injection of an immunotherapy, a mutant plasmid cDNA for the A1 adenosine receptor (A1 AR), targets the A1 AR in the dendritic cell. This mutant plasmid cDNA for the A1 AR reduced tumor volume in a dose- dependent manner that was sustained in the recovery period. For a six-week treatment phase this immunotherapy reduced tumor volume 35% for a low dose (50 µg), 47% for a midrange dose (100 µg) and 87% for a high dose (200 µg) versus diluent alone (p 0.002). For a two-week recovery phase versus diluent alone this immunotherapy produced a dose-dependent response reducing tumor volume 52% for the low dose (p 0.02), 77% for the midrange dose (p 0.002) and 93% for the high dose (p 0.003). These results support that a mutant plasmid cDNA for the A1 AR produces therapeutic efficacy in a xenograft model of PC3 human prostate cancer. It is expected that this approach to the treatment of prostate cancer will improve mortality and quality of life by treating both local and systemic disease while avoiding side effects associated with surgical and nonsurgical approaches.
Keywords: Immunotherapy; Intratumor; A1 Adenosine Receptors
Introduction
The American Cancer Society (ACS) estimates that 268,490 new cases of prostate cancer will occur in 2022. Moreover, the ACS estimates that 34,500 men will die from prostate cancer in 2022.
Prostate cancer remains the third leading cause of cancer death in men [1]. Presently surgery, radiation therapy (RT), and proton beam therapy are used to treat prostate cancer. Moreover, chemotherapy, hormonal therapy, cryosurgery, and high intensity focused ultrasound (HIFU) may be used depending on the clinical presentation. These treatments are not without side effects.
Based on the microscopic architecture and appearance of cells in prostate biopsies, Gleason patterns and scores are determined [1]. Taken together with the prostate specific antigen (PSA) level and clinical stage, i.e. with or without lymph node involvement or metastases, clinicians stratify patients into low, intermediate and high risk.
Patients with low risk and some with intermediate risk may opt for active surveillance (AS), RT or brachytherapy over surgery understanding that surgery is associated with erectile dysfunction (81%) and bowel and urinary incontinence (12 and 17%, respectively) which reduce the overall quality of life [1]. Radiation treatment may be recommended for a primary treatment or adjunctive treatment following surgery in patients with positive resection margins. However, RT is associated with erectile dysfunction (66%), as well as bowel and urinary incontinence (6% and 4%, respectively), albeit lower than that of surgery. The overall cause for mortality for surgery ranges from 10-57% while the mortality rate for prostate cancer ranges 0.9 – 6% versus 10 – 71% and 0.7 – 28% mortality for overall cause and prostate cancer, respectively for AS (1). The mortality rate for RT is 10% and 0.7%, for overall and prostate cancer, respectively
In patients with low to intermediate risk nonsurgical therapies, including brachytherapy and ablative therapies may be recommended. Ablative therapies may be used as a primary treatment or as a salvage treatment for patients that fail RT. Ablative therapies include high-intensity focused ultrasound (HIFU), interstitial laser ablation therapy (ILAT), vascular targeted photodynamic therapy (PDT) and cryotherapy (CT). Adverse effects of HIFU include erectile dysfunction and bladder outlet obstruction.
Other nonsurgical treatments for prostate cancer include androgen-deprivation therapy (ADT), chemotherapy and emerging immunotherapies [2]. Androgen-deprivation therapy is recommended for advanced disease. ADT may slow tumor progression and alleviate symptoms of advanced disease. The goal of ADT therapy is to achieve maximum androgen blockade with castrate levels of testosterone until castrate–resistant disease emerges. Chemotherapy and emerging immunotherapies are used in patients with castrate-resistant disease and advanced prostate cancer. Side effects of chemotherapeutic agents such as abiraterone acetate, cabazitaxel, denosumab, and enzalutamide include hypertension, edema, hypokalemia, neutropenia, diarrhea, hypocalcemia and seizures [1].
Dendritic cells are key players in the biology of cancer, including prostate cancer. It is previously reported that A1 ARs are downregulated in dendritic cells matured with lipopolysaccharide [3]. Also, it was previously reported that lipopolysaccharide binds to and activates A1 ARs [4]. Certain amino acids for the 326 amino acid cDNA for the A1 AR are vulnerable to phosphorylation, desensitization, and down-regulation of A1 ARs resulting in a defective protein and immunosuppressed dendritic cell. A defective cDNA for the A1 AR may render the dendritic cell ineffective in the uptake, processing or presentation of tumor antigen to naïve T cells in the tumor microenvironment.
To test this hypothesis in a proof of concept study in Athymic nude mice xenograph model of PC3 human prostate cancer cells, a midrange dose for the mutant plasmid cDNA for the A1 AR, following injection twice weekly for six weeks directly into the tumor, reduced tumor volume by 66% (data not shown). For this mutant plasmid cDNA, a single amino acid was substituted for the wild type cDNA amino acid to promote resistance to phosphorylation and desensitization of the A1 AR. The objective of the present definitive study is to demonstrate a dose-dependent response and sustained efficacy in this mouse model of human prostate carcinoma with this mutant plasmid cDNA for the A1 AR.
This present study demonstrates a dose-dependent and sustained therapeutic efficacy of a novel immunotherapy for prostate cancer following intratumor administration. It is expected that this approach to the treatment of prostate cancer will improve mortality and quality of life by treating both local and systemic disease while avoiding side effects associated with surgical and nonsurgical approaches.
Materials and Methods
Synthesis of the mouse mutant A1 adenosine receptor plasmid cDNA (Life Technologies, Corp, Carlsbad, CA)
The synthetic gene, mouse mutant A1 adenosine receptor plasmid cDNA, mouse Protein X, was assembled from synthetic
oligonucleotides and/or PCR products. The fragment was inserted into pMARQ (AmpR). The plasmid DNA was purified from transformed bacteria and concentration determined by UV spectroscopy. The final construct was verified by sequencing. The sequence identity within the insertion sites was 100%.
Formulation of plasmid DNA (pDNA) for mouse mutant A1 adenosine receptor – Lipid Nanoparticle (LNP) (Avanti Polar Lipids, Alabaster, AL)
Lipids were blended in solvent and dried to a film. Lipid film was then placed under vacuum overnight to remove residual solvent. Lipid film was dissolved in ethanol to produce the organic phase. Plasmid DNA was received in phosphate-buffered saline. Mouse IFNγ was dissolved in phosphate-buffered saline and combined with mouse plasmid DNA to make up the aqueous phase. The aqueous and organic phases were combined at controlled flow rates via microfluidics to produce lipid nanoparticles. Lipid nanoparticles were concentrated via tangential flow filtration, and then diafiltered fifteen times against phosphate-buffered saline. Mouse anti-CD11c antibody was conjugated to lipid nanoparticles by incubating at 2 °C for approximately 18 hours. Conjugated lipid nanoparticles were then concentrated using centrifugal filtration. The resultant product was packaged under nitrogen into amber vials.
Treatment in a Xenograft Model of PC3 Tumors (human prostate carcinoma) with a mutant mouse pDNA for the A1 adenosine receptor in Athymic Nude Mice (Washington Biotechnology, Inc., Baltimore, MD)
Animals
All procedures and animal care are approved by the Animal Care Committee of Washington Biotechnology, Inc. Eighty-two Athymic nude mice (ENVIGO, male, 5-6 weeks’ old were used in this study. All mice were ear tagged for identification purposes. Upon arrival, animals were examined to ensure that they were healthy. The animals were housed in autoclaved solid floor polycarbonate cages. Housing and sanitation were performed in accordance with Washington Biotechnology standard operating procedures. All animal handling was performed in a laminar flow hood. Animals were housed in filter-topped cages within a Hepa filtered clean room.
Cell culture and Implantation
PC3 cell line for human prostate cancer was obtained from American Type Culture Collection (Manassas, VA). The cells were cultured in 75 cm2 flask containing Eagle's Minimum Essential Medium supplemented with 10% fetal bovine calf serum (FBS) and incubated at 37°C in humidified atmosphere of 5% CO2. As cells became 80% confluent, cultures were expanded to 150 cm 2 flasks, and expanded further until sufficient cells were available for injection. Cancer cells were subcutaneously injected into right flank, 10 million cells/each mouse of PC3 cells.
Treatment Protocol
Animals were assigned to 7 study groups with 10 mice in each group based upon tumor volume. Dosing is shown in Table 1. The effect of treatments on animal body weight and tumor volume were monitored. Xenograft tumors were measured three times a week with a digital caliper. Tumor volumes were calculated using the formula: Tumor Volume = length x width x width x ½.
Treatment groups: Group 1, LNP, vehicle control; Group 2 low dose mouse mutant plasmid cDNA for A1 AR; Group 3 mid-range dose mouse mutant plasmid cDNA for A1 AR; Group 4 high dose mouse mutant plasmid cDNA for A1 AR; Group 5 mouse wild type plasmid cDNA for A1 AR; Group 6 mouse INFγ; Group 7 Paclitaxel chemotherapeutic agent as positive control.
Treatment groups 1 - 6 are treated with intratumor injections. Treatment groups 1 – 6 are treated with mouse anti-CD11c antibody, a biomarker for the mouse dendritic cell. Treatment groups 2 – 5 are treated with mouse INFγ (275 ng/injection volume). Treatment group 6 is treated with mouse INFγ (275 ng/injection volume)
The intratumor injection volume for groups (1 – 6) is 27 µl.
A1 AR, A1 adenosine receptor; LNP, lipid nanoparticle; INFγ, interferon gamma
Statistical Analysis
Tumor sizes and body weights were analyzed using Student's t-test. P values <0.05 were considered as statistically significant.
Results
Effect of Treatment on Mouse Weight (g)
There was no loss of body weight in treatment groups compared with dose started (Day 0) (Data not shown).
Effect of treatments on tumor volume
Treatment Period
1. Effect of Treatment on Mouse Weight (g):
There was no body loss in treatment groups compared with dose started (Day 0).
2. Effect of treatments on tumor volume size
A. Treatment Period
1) Group 2: There was no statistical difference (p 0.232) (last day) in tumor sizes between Group 1 (vehicle group) and Group 2. Tumor volume reduction was 35% and one tumor disappeared for Group 2.
2) Group 3: There was no statistical difference (p 0.098) in tumor size between Group 1 and Group 3 (mid dose mupDNA). Tumor volume reduction was 47% and three tumors disappeared in Group 3.
3) Group 4: There was statistical difference in tumor size between Group 1 and Group 4 (high dose mupDNA) (p 0.002). Tumor reduction was 87% and five tumors disappeared in Group 4. Moreover, versus Group 5 (wild type pDNA) there was a 71% reduction in tumor volume for Group 4. There is a dose-response for tumor volume reduction of 35%, 47% and 87% for Groups 2, 3, and 4, respectively.
4) Group 5: There was statistical difference in tumor size in Group 5 versus Group 1 (p 0.03). Tumor reduction was 54% and four tumors disappeared. Compared to Group 5 there was no statistical difference for any treatment group except Group 1 (p 0.03) and Group 7 (p 0.001)
5) Group 6: There was no significant difference in tumor size between Group 1 and Group 6 (INFγ) (p 0.057). Tumor reduction for Group 6 was 47% and two tumors disappeared.
6) Group 7: There was statistical difference in tumor sizes between Group 1 and Group 7 (Paclitaxel) (p 0.001). Tumor reduction was 100% and all tumors disappeared in Group 7.
B. Recovery Period
1) Group 2: There was statistical difference (p 0.022) between Group 1 and Group 2 in tumor size. Tumor reduction was 52% for Group 2. Moreover, versus Group 5 the there was an 8% reduction in tumor volume for Group 2.
2) Group 3: There was a statistical difference between Group 1 and Group 3 (p 0.002). Tumor reduction was 77% for Group 3. Moreover, versus Group 5 there is a 56% tumor reduction in tumor volume for Group 3.
3) Group 4: There is a statistical difference (p 0.003) between Group 1 and Group 4. Tumor reduction was 93%. Moreover, versus Group 5 there was an 86% reduction in tumor volume for Group 4. There is a dose-response for tumor volume reduction of 52%, 77% and 93% for Groups 2, 3, and 4, respectively.
4) Group 5: There is no significant difference (p 0.052) between Group 1 and Group 5 or Group 5 and Group 2 (p 0.848) and Group 3 (p 0.164). There is a statistical difference (p 0.035) between Group 4 and Group 5. Tumor volume for Group 4 is 124 ± 64 versus that for Group 5 is 904 ± 290 (p 0.04). Compared to Group 5 there is no statistical difference for any treatment group except Group 4 (p 0.04) and Group 7 (p 0.02). Only Group 4 and Group 7 produced sustained reductions in tumor volume at 2 weeks.
5) Group 6: There is no significant difference (p 0.173) between Group 6 and Group 1. Tumor reduction was 32% for Group 6. Moreover, there is no statistical significance (p 0.538) in tumor reduction between Group 6 and Group 5.
6) Group 7: There is a statistical difference between Group 1 and Group 7 (p 0.004). Tumor reduction was 100% for Group 7.
Conclusion
There is a dose response for Groups 2, 3 and 4, low, mid-range, high dose mouse mutant plasmid cDNA A1 AR, respectively, for both the treatment and recovery periods. There is a sustained tumor reduction in the recovery period for Groups 2, 3 and 4. Statistical analysis shows that the mouse mutant plasmid cDNA A1 AR has antitumor efficacy with a dose-response in a xenograft model of PC 3 human prostate carcinoma in Athymic nude mice. No toxicity was observed in mice in Groups 2, 3, and 4 treated with the mouse mutant plasmid cDNA A1 AR.
Although Group 5 (mouse wild type pDNA A1 AR) produced a modest reduction in tumor volume during the treatment period this effect was not sustained during the recovery period. Statistical analysis suggests that the mouse wild type plasmid cDNA A1 AR produced modest antitumor efficacy in a xenograft model of PC 3 human prostate carcinoma in Athymic Nude Mice. This effect was significantly reduced compared to the antitumor activity of the high dose (Group 4) for the mouse mutant plasmid cDNA A1 AR.
Group 6 (mouse INFγ) did not produce a reduction in tumor volume in the treatment or recovery periods. Statistical analysis shows INFγ has no significant antitumor efficacy in xenograft model of PC 3 human prostate carcinoma in Athymic nude mice.
Group 7 (Paclitaxel) produced a significant reduction in tumor volume that was sustained in the recovery period of 100%. Statistical analysis shows Paclitaxel as a positive control works well in a xenograft model of PC3 human prostate carcinoma in Athymic nude mice.
There was no loss of body weight for any treatment group.
There was no toxicity observed in mice in Group 1 treated with vehicle, in mice in Group 5 treated with the mouse wild type plasmid cDNA A1 AR, in mice in Group 6 treated with mouse INFγ, or in mice in Group 7 treated with Paclitaxel.
Dicussion
The American Cancer Society (ACS) estimates that 268,490 new cases of prostate cancer will occur in 2022. Moreover, the ACS estimates that 34,500 men will die from prostate cancer in 2022.
Prostate cancer remains the third leading cause of cancer death in men [1]. Globally a total of 1,414,259 new cases of prostate cancer and 375,304 deaths were reported in in 174 countries in 2020 [5]. In 112 countries prostate cancer was the most frequently diagnosed cancer. And, it was the leading cause of cancer death in 48 countries.
Many treatments for prostate cancer including surgery, RT, ADT, and chemotherapeutic agents cause serious side effects and reduce the quality of life for patients with early stage and advanced prostate cancer.
Based on an understanding of the mechanisms by which tumors evade destruction by the immune system, a number of immunotherapies have been developed. These mechanisms of evasion by tumors include down modulation of components of antigen processing and presentation, recruitment of suppressor immune cells, such as regulatory T cells, myeloid suppressor cells, and tumor associated macrophages, production of soluble factors which produce immune suppression, such as TGF-β and IL-10, and upregulation of ligands for receptors that down modulate tumor infiltrating lymphocytes, such as programmed cell death ligand-1 (PD-L1) [6,7]. As such therapeutics, including antibodies to immune checkpoints, such as PD-1 and cytotoxic T cell lymphocyte antigen-4 (CTLA-4), are now FDA approved for the treatment of solid tumors.
Other immunomodulating factors include IFNs and cytokines, IL-2, IL-15, IL-21, and IL-7 [5]. IFN-α- 2b is approved as an adjuvant treatment for melanoma. And, IL-2 has been FDA approved for the treatment of metastatic renal cell carcinoma. Another approach to modulating the immune system is with the use of chimeric antigens for the T cell receptor, CAR T-cell adoptive immunotherapy. This approach has shown promising results in clinical trials [6,7]. In prostate cancer, the use of a viral- based immunotherapy encoding modified forms of prostate specific antigen (PSA) along with three co-stimulatory molecules, CD80, intercellular adhesion molecule-1 (ICAM-1) and lymphocyte function-associated antigen-3 (LFA-3) showed promising results in early clinical trials; however, it failed to meet its primary endpoint for overall survival in Phase III clinical trials [8].
A number of intratumor immunotherapies, including toll receptor agonists and oncolytic viral therapies, are in clinical trials and show promising anti-tumor activity in solid tumors with tolerable toxicities [9, 10]. Talimorgene laherparepvec (T-VEC), a genetically modified herpes simplex virus type 1, is FDA approved for the treatment of unresectable melanoma.
The immunotherapy, mutant plasmid cDNA for the A1 AR, described in this current study administered into the tumor, xenograft of PC3 human prostate carcinoma, in Athymic mice, demonstrated a dose-dependent and sustained therapeutic efficacy and had no adverse effects. It is expected that this mutant plasmid cDNA for the A1 AR was taken up by resident dendritic cells (DCs) since a biomarker for the mouse dendritic cell, anti-CD11c antibody, was conjugated to the lipid nanoparticle (LNP). Dendritic cells then activated naive T cells to kill tumor cells. It is expected that these educated T cells would hone to local lymph nodes activating additional T killer cells which would then act systemically to kill tumor cells. As such, this therapy should be effective in treating both local and systemic disease for solid tumors. Moreover, this therapy should be effective in treating both early stage and advanced prostate cancer.
Adenosine is an important signaling molecule in cancer [11]. It is secreted by both cancer and immune cells in the tumor microenvironment. It exerts its immunomodulatory effects in cancer by acting on A1, A2a, A2b and A3 ARs expressed on a number of different immune cells, including macrophages, myeloid-derived suppressor cells, dendritic cells, natural killer cells, T cells and regulatory T cells, all of which play an important role in the pathogenesis of cancer. Therapies targeting adenosine receptors for the treatment of cancer have been in clinical trials, and include an A2b AR antagonist, A2a AR antagonists and an A3a AR agonist [12, 13].
A1 adenosine receptors are expressed on immature DCs and induce chemotaxis, calcium transients and actin polymerization [3]. However, A1 ARs are down-regulated on lipopolysaccharide (LPS) induced maturation of DCs. Lipopolysaccharide binds to and activates A1 ARs [4]. It is possible that LPS induces desensitization of A1 ARs on mature DCs, thus rendering the mature DCs immunosuppressed to the activating effect of adenosine on A1 ARs in the tumor microenvironment.
The A1 adenosine receptor is a 326 amino acid protein. Some A1 adenosine receptor amino acids are at risk of phosphorylation, down regulation and desensitization rendering the protein incompetent Desensitization of A1 ARs occurs following exposure to an A1 AR agonist [14, 15]. This desensitization was reversed after treatment with protein phosphatases and was β-arrestin 1/ ERK 1/2 MAP kinase pathway dependent.
In the current study, treatment with a plasmid mutant cDNA wherein there is a substitution for a single amino acid for the A1 AR that renders the cDNA resistant to the effect of phosphorylation and desensitization, produces a dose-dependent and sustained highly effective anti-tumor effect in a xenograft model of PC3 human prostate carcinoma in Athymic nude mice.
The mechanism of action for this anti-tumor effect of the mutant plasmid cDNA for the A1 AR in mature DCs is not known. It is possible that the A1 AR is pivotal for the uptake, processing and/or presentation of the tumor antigen to naive T cells. These are areas for future investigations.
Conflict of Interest
Constance N Wilson is Chief Scientific Officer and owns equity in Endacea, Inc.
- Litwin MS, Hung JT (2017) The diagnosis and treatment of prostate cancer: a review. JAMA 317: 2532-42.
- Evans AJ (2018) Treatment effects in prostate cancer. Mod Pathol 31: S110-21.
- Panther E, Idzko M, Herouy Y, Rheinen H, Gebicke-Haerter PJ, Mrowietz U et al.(2001) FASEB J 15: 1963-70.
- Wilson CN, Batra VK (2002) Lipopolysaccharide binds to and activates A(1) adenosine receptors on human pulmonary artery endothelial cells. J Endotoxin Res 8: 263-71.
- Wang L, Bin L, He M, Wang Y, Wang Z, Lingbin D (2022) Prostate cancer incidence and mortality: Global status and temporal trends in 89 countries from 2000 to 2019. Front Public Health 10: 1-18.
- Yang Y (2015) Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest 125: 3335-7.
- Velcheti V, Schalper K (2016) Basic overview of current immunotherapy approaches in cancer. ASCO Educational Book 298-308.
- Slovin SF (2020) Immunotherapy for prostate cancer – treatments for the “lethal” phenotype. Urol Clin North Am 47: 469-74.
- Hong WX, Haebe S, Lee AS, Westphalen, Norton JA, Jiang W et al. (2020) Intratumoral immunotherapy for early-stage solid tumors. Clin Cancer Res 26: 3091-9.
- Hamid O, Ismail r, Puzanov I (2020) Intratumoral immunotherapy – update 2019. The Oncologist 25: 423-38.
- Kumar V (2013) Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go? Purinergic Signaling 9: 145-65.
- Sek K, Molck C, Stewart GD, Kats L, Darcy PK, Beavis PA (2018) Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci 19: 1-23.
- Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P et al. (2019) Targeting adenosine in cancer immunotherapy to enhance T–cell function. Front Immunol 10: 1- 30.
- Nie Z, Mei Y, Ramkumar V (1997) Short term desensitization of the A1 adenosine receptors in DDT1 MF-2 cells. Mol Pharmacol 52: 456-64.
- Jojoo S, Mukherjea D, Kumar S, Sheth S, Kaur T, Rybak LP et al. (2009) Role of β-arrestin 1/ERK MAP kinase pathway in regulating adenosine A1 receptor desensitization and recovery. Am J Physiol Cell Physiol 298: 56-65.
FIGURE 1
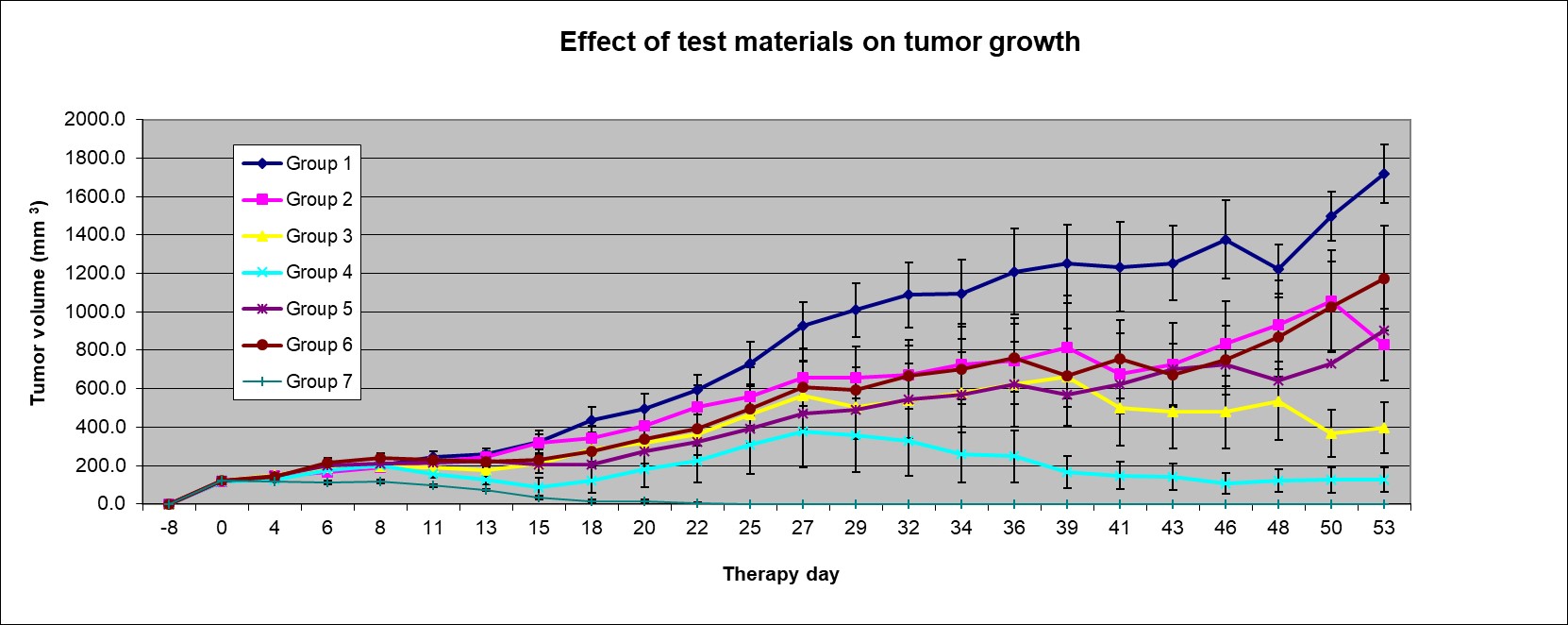
Figure1: Effect of test materials on tumor growth (mm3) was measured in a xenograph model of human PC-3 prostate cancer in athymic nude mice during a 6-week treatment period and a 2-week follow-up period Effect of Test Materials on Tumor Growth
FIGURE 2
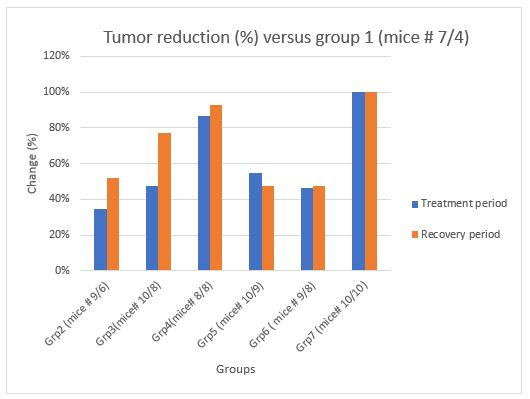
Figure2: Tumor reduction (% change) in the treatment groups 2 - 7 versus group 1 (diluent control) was measured in a xenograph model of human PC-3 prostate cancer in athymic nude mice during a 6-week treatment period and a 2-week recovery period. Grp denotes Group Tumor Reduction (%) Versus Group 1
FIGURE 3
Figure3: Plasmid Map
Tables at a glance
Figures at a glance